
More Information
Submitted: July 06, 2022 | Approved: July 11, 2022 | Published: July 12, 2022
How to cite this article: Adediran EO. Pefloxacin and its derivative, novel inhibitors of the SARS-CoV-2 Main protease (3CLpro) and their pharmacokinetics prediction: An in silico analysis. Arch Pharm Pharma Sci. 2022; 6: 013-018.
DOI: 10.29328/journal.apps.1001030
Copyright License: © 2022 Adediran EO. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Pefloxacin; 1-ethyl-6-fluro-7-(4(Nsubstitutedcarbamoylmethylphenyl)piperazyn-1-yl)-4-oxoquinoline-3-carboxylic acid; SARS-CoV2; Main-protease(3CLpro); Docking

Pefloxacin and its derivative, novel inhibitors of the SARS-CoV-2 Main protease (3CLpro) and their pharmacokinetics prediction: An in silico analysis
Emmanuel Oluwaseun Adediran*
Department of Chemistry, Georgia State University, Atlanta 30303, USA
*Address for Correspondence: Emmanuel Oluwaseun Adediran, Department of Chemistry, Georgia State University, Atlanta 30303, USA, Email: [email protected]; [email protected]
For over two years, COVID-19 pandemic has been a major global health concern and threat to human life. In the SARS-CoV2 macromolecules, the 3-chymotrypsin like protease (3CLpro or main protease) has been identified to be crucial and essential for viral survival, processing of the viral polyproteins and has been explored as a target in COVID-19 drug discovery.
Although vaccines and other various inhibitors have been designed and launched, the emergence of the variant of this virus has put an unrelenting effort of researchers to this end. Also, the high cost of manufacturing these molecules coupled with the occurrence of drug resistance is a concern.
Herein, Pefloxacin and its derivative for the first time were screened for their inhibitory activity against the SARS-CoV2 main protease through in silico analysis and their pharmacokinetic properties were evaluated. Interestingly, from the docking results, they both bind with high affinity at the active site of the protein. Moreover, they showed excellent pharmacokinetic and drug - likeness properties. Derivatization of Pefloxacin at the C7 position prevents its blood-brain barrier permeability.
Overall, the dual antibacterial and potential antiviral activities of these two molecules make them promising drug candidates for COVID-19 management.
Before now, fluoroquinolones and other synthetic analogs have been approved for their antibacterial activities and efficacy against respiratory tract infection [1,2]. Their mechanism of action is based on the ability to inhibit DNA Gyrase-topoisomerase II and topoisomerase IV - the enzyme crucial for DNA replication and synthesis [1]. A previous study has reported the biological activities of fluoroquinolones against varicella-zoster virus, and cytomegalovirus [3]. Additionally, clinical data have exposed the activities of fluoroquinolone and quinolone-based drugs against DNA and RNA viral infections [4].
The fluoroquinolone skeletal structure consists of a carboxylic group at C-3 and a bulky substituent at the C-7 position and the N1 position of the quinolone moiety. These key structural features and modifications have been identified and reported to enhance their antiviral activities and pharmacokinetic properties [5]. In line with this background, ciprofloxacin and his chalcone derivatives have been reported to inhibit the SARS-CoV2 main protease via molecular docking studies and in vitro studies [6]. Also, Moxifloxacin was reported to interact with the SARS-CoV2 protease via the preliminary in silico analysis [7]. In the same perspective, a Library of Quinolones-based agents were used to probe the different macromolecular targets of the SARS-CoV2 and several inhibitors were reported [8].
The coronavirus disease caused by the several acute syndrome coronavirus 2(SARS-CoV2) originated from Wuhan in china and displays symptoms ranging from fever, and dry cough to shortness of breath [9]. In the SARS-CoV2 macromolecules, 3-chymotrypsin like protease (3CLpro or main protease) is essential for the viral life cycle [10]. This enzyme plays a crucial role in the processing of the viral polyproteins and has been explored as a target in COVID-19 drug discovery [11]. According to crystallographic data, amino acid HIS 41, HIS 164, MET 49, MET 165, THR 190 and GLY 143 play a very crucial role in the stability of the ligand - Mpro complexes and are key amino acid residues in the active site [12,13].
Although vaccines and other various inhibitors have been designed as shown in Table 1, the emergence of the variant of this virus has put an unrelenting effort of researchers to this end. Also, the high cost of manufacturing these molecules is a disadvantage to developing countries. Unfortunately, the presence of comorbid conditions of mixed viral and bacterial infections coupled with the emergence of drug resistance demands more effort in drug discovery and repurposing.
| Table 1: Reported inhibitors across all macromolecular targets of SARS-CoV-2 and their pharmacological classes. | |||
| Compounds | Pharmacological class | Targets inhibited | References |
| Grl-0240-20 | Benzothiazolyl inhibitor | Main protease | [14] |
| Quercetin, Apigenin |
Natural products/Flavonoids | Main protease | [15] |
| Rutin Lopinovir Emetine Hesperidine Ritonavir |
Natural products Antiviral Antiemetic Natural product Antiviral |
Main protease | [16] |
| C-1 cid 11170714 | Marine natural product | Main protease | [17] |
| Calycin and Rhizocarpic acid | Lichen natural product | Main protease | [18] |
| Lopinavir Darunavir Z31792168 (2-cyclohexyl-N-pyridin-3-yl-ethanamide) |
Antiviral/alpha-ketoamide | Main protease | [19] |
| (Oolonghomobisflavan-A, Theasinensin-D, and Theaflavin-3-O-gallate) | Natural products | Main protease | [20] |
| Repaglinide Canagloflozin Glimepiride Linagliptin Glipizide |
Antidiabetic drugs | Main protease | [21] |
| (E)-N-(4-cyanobenzylidene)-6-fluoro-3-hydroxypyrazine-2-carboxamide | Anti-influenza / Antiviral |
RNA-dependent RNA polymerase | [22] |
| Cepharanthine Nelfinavir |
Anti-inflammatory drug Antiviral drug |
Viral entry inhibitor | [23] |
| Compounds 29 and 34 | Synthesized indole-based compounds | Dual inhibitor Both main protein and spike protein |
[24] |
| Ccg-50014 | Thiazolidine derivative | Main protease | [25] |
| Calpeptin | Small molecules | Spike protein | [26] |
| 14-deoxy-11,12-didehydroandrographolide, Costunolide, Germacranolide and Hetisinon | Natural Product | Spike protein | [27] |
| Lead compound 46 | Ketoamide derivative | Main protease | [28] |
Herein, for the first time, we probe and investigate the interaction of the SARS-CoV2 main protease with Pefloxacin and its derivate and studied the ligand-protein binding using in silico molecular docking techniques. We hypothesize that a structural modification at the C-7 position could modulate the antiviral activity of Pefloxacin and improve its pharmacokinetic properties.
Ligand preparation
Several compounds have shown their various antiviral activities from docking studies [29]. In this study, The PDB format of Pefloxacin and Hydroxychloroquine are retrieved from the PubChem database.Derivativeofpefloxacin-(1-ethyl-6-fluro-7-(4-(Nsubstitutedcarbamoylmethylphenyl)piperazyn-1-yl)-4-oxoquinoline-3-carboxylic acid was drawn and optimized with MolView and finally saved as a PDB format.
Following energy minimization and optimization of the ligands [30], they were all converted to PDBQT format using the graphical user interface version of PyRx virtual screening tool-python prescription 0.8.
Protein preparation
The 3D co-crystallized ligand of SARS-CoV-2 3CLpro (PDB ID: 6LU7) with the resolution of 2.16 Å was retrieved from the RCSB PDB database (http://www.rcsb.org/pdb). The protein was extracted to expose the active site and water of crystallization was removed [31]. Polar hydrogens were added and the protein was further optimized by the Autodock tool and Biovia Discovery Studio Visualizer before the docking process [32].
Molecular docking
The virtual screening of the compounds was done with hydroxychloroquine as the control using Pyrx software by autodock wizard as the engine for docking [33,34]. The configuration file for the grid parameters was generated using Auto Grid engine in Pyrex. Predefined XYZ Center Coordinate of -13.7042, 14.7095,72.0248 respectively having amino acid residues in the active site of the protein were set using the Vina wizard. The ligands with the highest binding energy, preferred orientation and lowest RMSD were considered to be ligands with very high affinity. Then the ligand-protein interactions were visualized by Biovia Discovery Studio Visualizer and the plausible binding modes are predicted [35]. Furthermore, the compounds were compared to hydroxychloroquine as the control.
Drug-likeliness properties
The Swiss ADME Predictor (http://www.swissadme.ch/) was used to calculate the Lipinski rule parameters and drug-likeliness properties [36-39].The canonical smiles of ligands were retrieved and entry was made into the server. These properties include molecular weight, number of hydrogen bond acceptor, number of hydrogen bond donor, topological polar surface area, and lipophilicity level. Human intestinal absorption, Blood brain barrier permeability were also investigated. Above mentioned parameters help to predict aqueous solubility and the ability of molecules to travel across the biological lipophilic membrane.
Virtual screening of the inhibitors of the SARS-CoV2 3CLpro (Main protease)
Table 2 molecular docking result reveals that pefloxacin and its analog for the first time have a very high binding affinity compared to hydroxychloroquine which is the control. Pefloxacin, Pefloxacin ananalognd Hydroxychloroquine have the binding energy of -7.9 kcal/mole, -7.6 kcal/mole and -6.0 kcal/mole respectively. The types of interactions and the amino acid residues in the active site that are involved in the interaction are shown in Figures 1-4. Pefloxacin interacts with the amino acid residues, THR 26, ARG 188 CYS 145, HIS 41, MET 49, MET 49. Pefloxacin derivative interact with the amino acid residues, THR 190, GLN 189, CYS 145, SER 144. Furthermore, Hydroxychloroquine interacts with amino acid residues- MET 49, GLN 189, ARG 188, MET 165, ASP 187, HIS 164, HIS 41, CYS 145, ASN 142, PHE 140, HIS 172.
| Table 2: Binding Energy of Pefloxacin, its analogue and hydroxychloroquine and the key amino acid residue of 3CLpro(Main protease) involved in the binding. | ||
| Compound | Energy score | Interacting residue |
| Pefloxacin Derivative | -7.6 kcal/mole | THR 190, GLN 189, CYS 145, SER 144 |
| Pefloxacin | -7.9 kcal/mole | THR 26, ARG 188 CYS 145, HIS 41, MET 49, MET 49 |
| Hydroxychloroquine | -6.0 kcal/mole | MET 49, GLN 189, ARG 188, MET 165, ASP 187, HIS 164, HIS 41, CYS 145, ASN 142,PHE 140, HIS 172 |
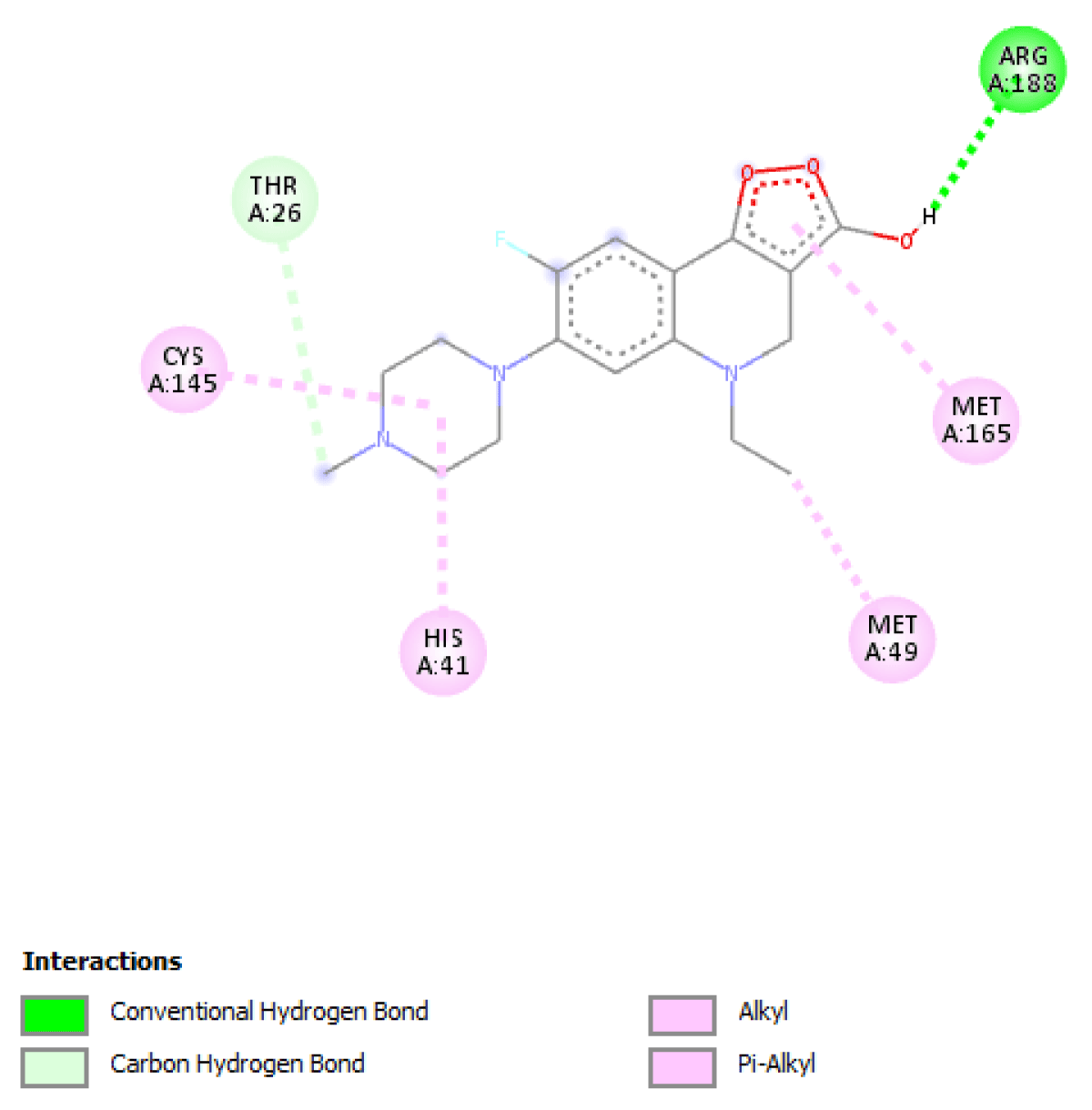
Figure 1: Interaction of Pefloxacin with 3CLpro(Main protease) and the key amino acid residues.
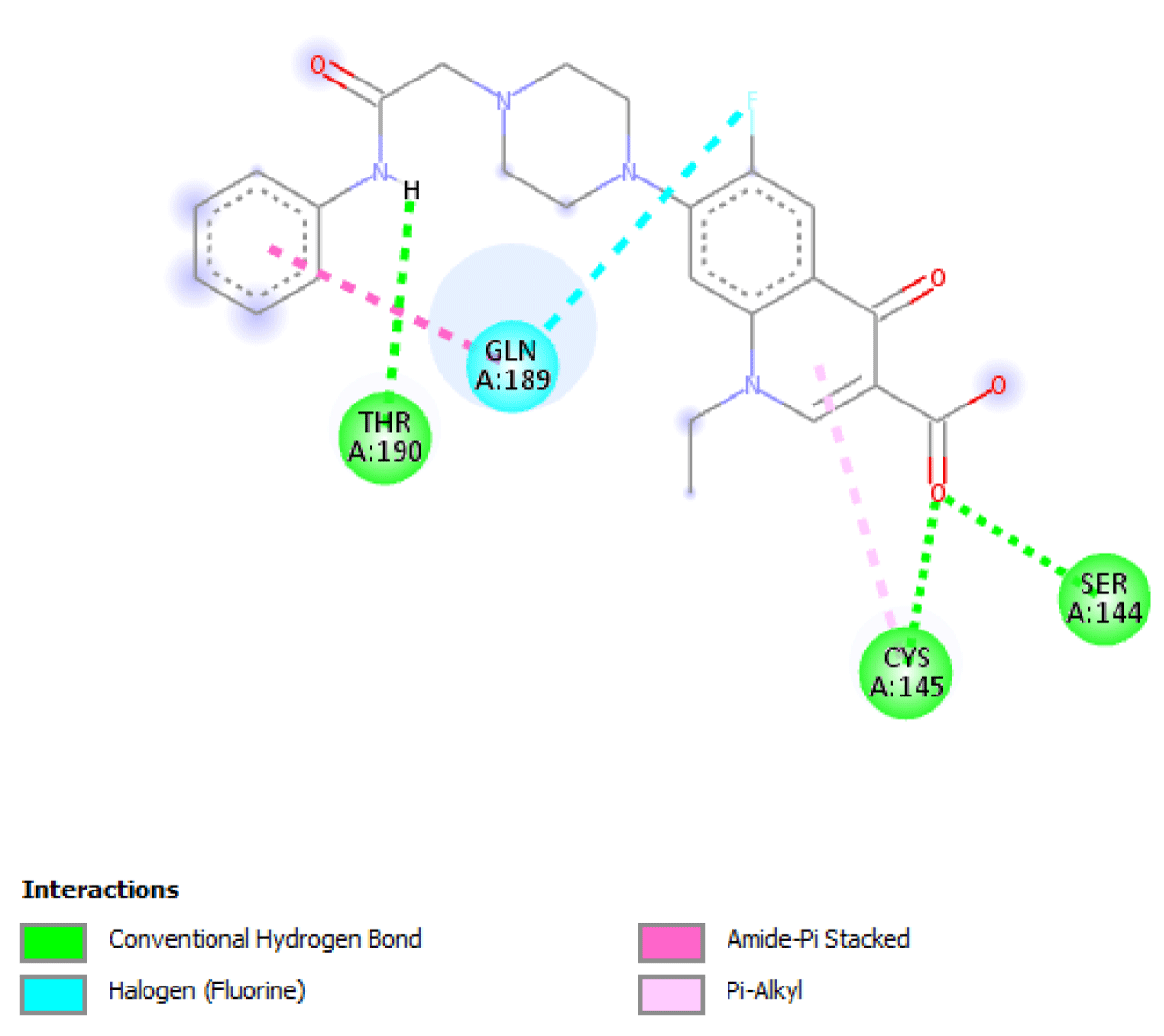
Figure 2: Interaction of 1-ethyl-6-fluro-7-(4-(N-substitutedcarbamoylmethylphenyl)piperazyn-1-yl)-4-oxoquinoline-3-carboxylic acid with 3CLpro(Main protease) and the key amino acid residues.
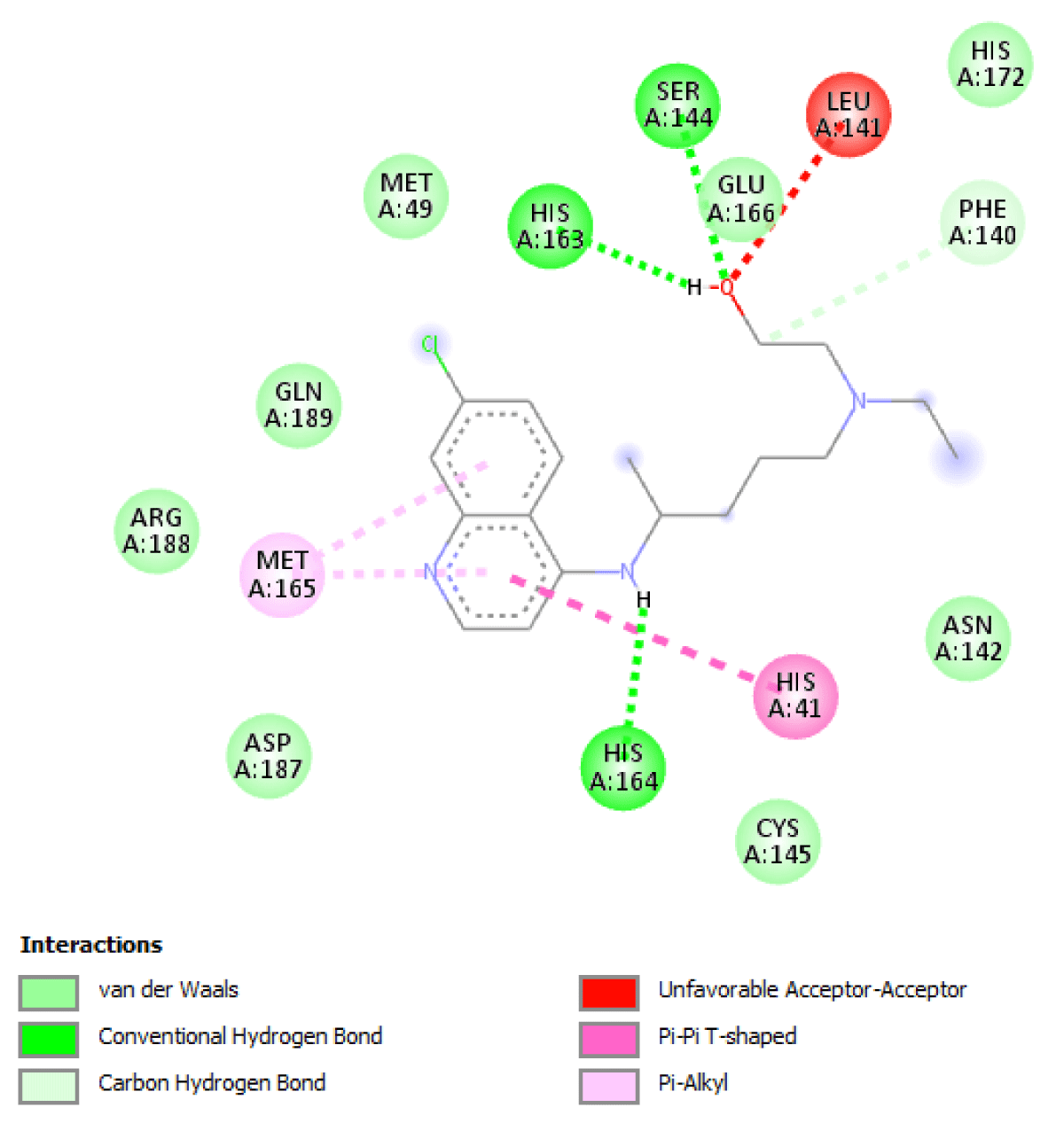
Figure 3: Interaction of Hydroxychloroquine with 3CLpro (Main protease) and the key amino acid residues.
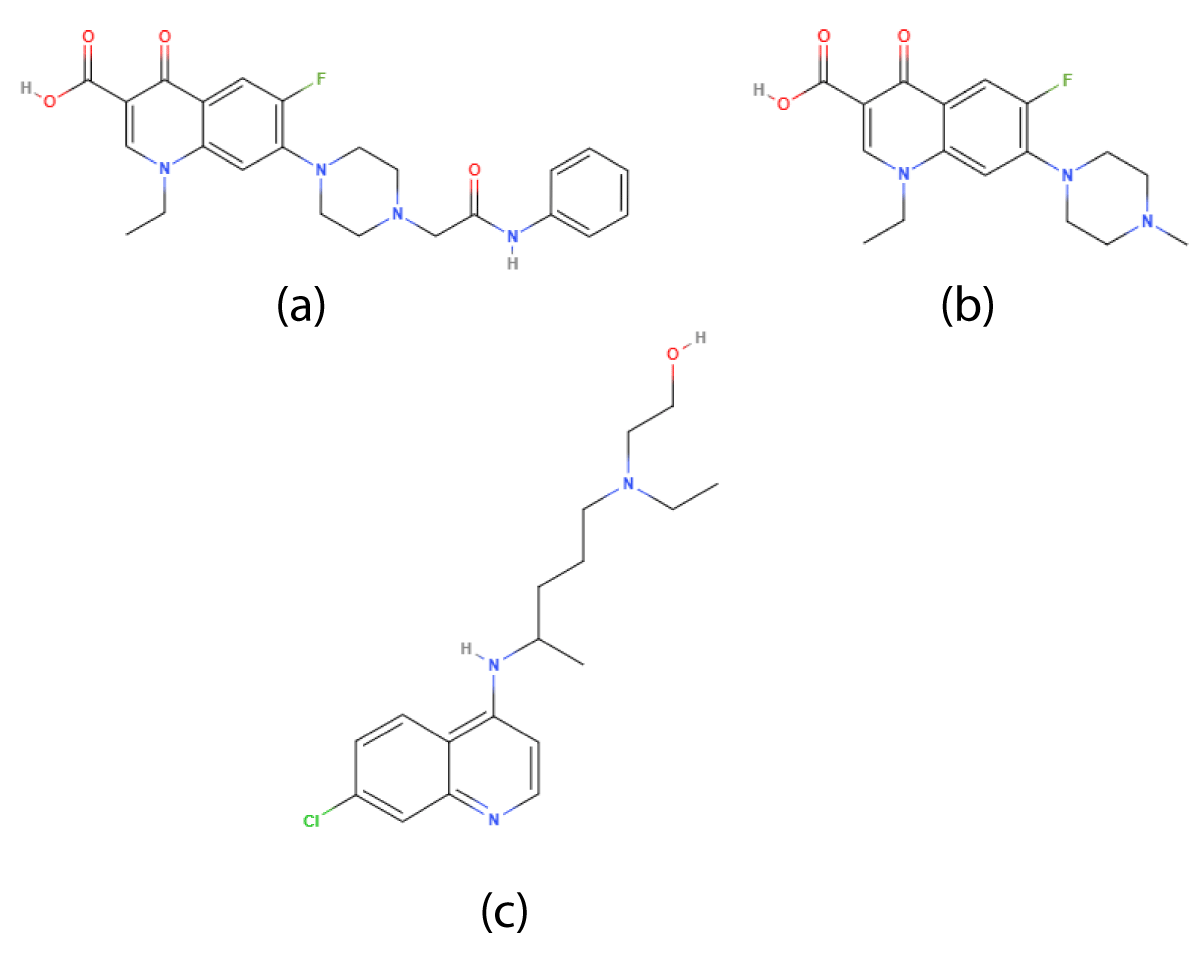
Figure 4: Chemical structure of (a)Derivative of perfloxacin-(1-ethyl-6-fluro-7-(4-(N-substitutedcarbamoylmethylphenyl)piperazyn-1-yl)-4-oxoquinoline-3-carboxylic acid (b)Pefloxacin (c)Hydroxychloroquine.
Drug likeliness properties and in silico ADME prediction
Properties such as molecular weight, number of hydrogen bond acceptor, number of hydrogen bond donor, topological polar surface area, lipophilicity level, Human intestinal absorption, Blood brain barrier permeability were calculated for Pefloxacin, its analogue and hydroxychloroquine and the result is as shown in Table 3.
The 3CLpro (main protease) of the SARS-CoV2 has been identified as one of the invaluable macromolecular targets in COVID-19 drug discovery. The protease plays a vital role in the viral replication, processing of the polyproteins that are translated from the RNA molecules. Moreover, the key amino acid residues in the active site that have been mapped out include, HIS 41, HIS 164, MET 49, MET 165, THR 190 and GLY 143. Also, several classes of drugs including fluoroquinolones have been screened for their ability to inhibit the main protease of the SARS-CoV2 [6]. Although vaccines and other various inhibitors have been designed and launched, the emergence of the variant of this virus has also put an unrelenting effort of researchers to this end. Also, the high cost of manufacturing these molecules coupled to the occurrence of drug resistance is a disadvantage to developing countries.
We screened pefloxacin and a structural modification of the position C-7 analogue for antiviral activity and compared them with hydroxychloroquine.
Our result revealed that for the first time that pefloxacin and its analog bind with a very high affinity and have a binding energy of -7.9 kcal/mole and -7.6 kcal/mole respectively compared to the control. Although Ciprofloxacin had earlier been reported to have a binding energy of -7.0 kcal/mole [7] Pefloxacin has a higher affinity when comparing the scores. The analysis of the ligand-protein interaction shows that pefloxacin and its analog show a very vital interaction with the key amino acid residue at the active site as shown in Table 2. Pefloxacin interact with the amino acid residues THR 26, ARG 188 CYS 145, HIS 41, MET 49, MET 49. Pefloxacin derivatives interact with the amino acid residues THR 190, GLN 189, CYS 145, SER 144. Furthermore, Hydroxychloroquine interacts with amino acid residues- MET 49, GLN 189, ARG 188, MET 165, ASP 187, HIS 164, HIS 41, CYS 145, ASN 142, PHE 140, HIS 172.
As shown in Table 3, Pefloxacin and its analog both has an hydrogen bond donor atoms and hydrogen bond acceptor atom less than 5 and 10 respectively. Pefloxacin and its structurally modified form both have a low TPSA (65.78 and 94.88) respectively coupled with a bioavailability scores greater than zero. These cumulatively suggest a higher possibility of antiviral activities. Interestingly, a structural modification at the C7 position impedes the blood brain barrier permeability of pefloxacin. iLog p - values less than 5 has presented in the table authenticate an ideal lipophilicity nature of the molecules. The two molecules have a high intestinal absorption ability. To our advantage, they are not classified as Pan-Assay Interference Compounds (PAINS) reflecting that they bind to specific biological sites instead of interacting with random targets.
| Table 3: Lipinski parameter of Pefloxacin, 1-ethyl-6-fluro-7-(4-(N-substitutedcarbamoylmethylphenyl) piperazyn-1-yl)-4-oxoquinoline-3-carboxylic acid and Hydroxychloroquine. | |||
| Lipinski Parameters | Pefloxacin derivate | Pefloxacin | Hydroxychloroquine |
| MW(g/mole) | 452.48 | 333.36 | 335.87 |
| HBA | 6 | 5 | 3 |
| HBD | 2 | 1 | 2 |
| Nrotb | 7 | 3 | 9 |
| TPSA(Ų) | 94.88 | 65.78 | 48.39 |
| ILogP | 2.69 | 2.23 | 3.58 |
| F | 0.55 | 0.55 | 0.55 |
| BBB | NO | YES | YES |
| CYP2C19 inhibitor | NO | NO | NO |
| HIA % | HIGH | HIGH | HIGH |
| Log S | -2.76 | -1.21 | -4.28 |
| PAINS | NO | NO | N0 |
| DRUG LIKELINESS | YES | YES | YES |
| BBB: Blood-Brain Barrier Permeability; CYP2CI9 inhibitor hepatotoxicity; F: Abbott bioavailability scores; MW: Molecular Weight; TPSA: Topological Polar Surface Area; PAINS: Pan-Assay Interference Compounds; Log S: Aqueous Solubility Scale; HIA: Human Intestinal Absorption; iLogP: n-octanol/water partition coefficient; HBD: Hydrogen Bond Donor; HBA: Hydrogen Bond Acceptor; Nrotb: Number of Rotatable Bonds. | |||
Favorably, the obtained calculations by the Swiss ADME predictor indicated that the two molecules meet the bioavailability requirement and are good drug candidates. However, the analog proposed is not commercially available but can be synthesized. It is also important to note that Pefloxacin as an example of fluoroquinolones are used as an antibiotic in the management of upper respiratory tract infections in cases of beta-lactam antibiotics resistance. Therefore, in this context, a dual mode of antiviral and anti-bacterial activity is possible to the great advantage of the COVID-19 patients.
For the first time, pefloxacin and its structural analog was screened for their inhibitory activity against the SARS-CoV2 main protease, and they both bind with high affinity compared to hydroxychloroquine via molecular docking approach. Docking pose are suggestive of the structural alteration and modification of antiviral activities. Providentially, these two drug molecules are potential drug candidate for COVID-19 management.
However, the derivatized compound needs to be synthesized. Also, in vitro, in vivo testing and further clinical trials need to be done on these molecules in order to elucidate their molecular mechanism and validate this novel finding.
- Correia S, Poeta P, Hébraud M, Capelo JL, Igrejas G. Mechanisms of quinolone action and resistance: where do we stand? J Med Microbiol. 2017 May;66(5):551-559. doi: 10.1099/jmm.0.000475. Epub 2017 May 12. PMID: 28504927.
- Suaifan GARY, Mohammed AAM. Fluoroquinolones structural and medicinal developments (2013-2018): Where are we now? Bioorg Med Chem. 2019 Jul 15;27(14):3005-3060. doi: 10.1016/j.bmc.2019.05.038. Epub 2019 May 31. Erratum in: Bioorg Med Chem. 2019 Nov 1;27(21):115072. PMID: 31182257.
- Richter S, Parolin C, Palumbo M, Palù G. Antiviral properties of quinolone-based drugs. Curr Drug Targets Infect Disord. 2004 Jun;4(2):111-6. doi: 10.2174/1568005043340920. PMID: 15180459.
- Ali SH, Chandraker A, DeCaprio JA. Inhibition of Simian virus 40 large T antigen helicase activity by fluoroquinolones. Antivir Ther. 2007;12(1):1-6. PMID: 17503741.
- Fedorowicz J, Sączewski J. Modifications of quinolones and fluoroquinolones: hybrid compounds and dual-action molecules. Monatsh Chem. 2018;149(7):1199-1245. doi: 10.1007/s00706-018-2215-x. Epub 2018 Jun 7. PMID: 29983452; PMCID: PMC6006264.
- Richter S, Parolin C, Palumbo M, Palù G. Antiviral properties of quinolone-based drugs. Curr Drug Targets Infect Disord. 2004 Jun;4(2):111-6. doi: 10.2174/1568005043340920. PMID: 15180459.
- Marciniec K, Beberok A, Pęcak P, Boryczka S, Wrześniok D. Ciprofloxacin and moxifloxacin could interact with SARS-CoV-2 protease: preliminary in silico analysis. Pharmacol Rep. 2020 Dec;72(6):1553-1561. doi: 10.1007/s43440-020-00169-0. Epub 2020 Oct 15. PMID: 33063271; PMCID: PMC7561236.
- Alexpandi R, De Mesquita JF, Pandian SK, Ravi AV. Quinolines-Based SARS-CoV-2 3CLpro and RdRp Inhibitors and Spike-RBD-ACE2 Inhibitor for Drug-Repurposing Against COVID-19: An in silico Analysis. Front Microbiol. 2020 Jul 23;11:1796. doi: 10.3389/fmicb.2020.01796. PMID: 32793181; PMCID: PMC7390959.
- GhebreyesusTA “WHO Director-General’s opening remarks at the media briefing on COVID-19 World Heal Organ”. 2020; 4.
- Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020 Apr 24;368(6489):409-412. doi: 10.1126/science.abb3405. Epub 2020 Mar 20. PMID: 32198291; PMCID: PMC7164518.
- Khan RJ, Jha RK, Amera GM, Jain M, Singh E, Pathak A, Singh RP, Muthukumaran J, Singh AK. Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2'-O-ribose methyltransferase. J Biomol Struct Dyn. 2021 May;39(8):2679-2692. doi: 10.1080/07391102.2020.1753577. Epub 2020 Apr 20. PMID: 32266873; PMCID: PMC7189412.
- Fearon D, Powell AJ, Douangamath A, Owen CD, Wild C, Krojer T, et al. PanDDA analysis group deposition—crystal structure of COVID-19 main protease in complex with Z1220452176. 2020.
- Liu X, Zhang B, Jin Z, Yang H, Rao Z. The crystal structure of COVID-19 main protease in complex with an inhibitor N3. 2020. https://doi.org/10.2210/pdb6LU7/pdb.
- Santana MVS, Silva-Jr FP. De novo design and bioactivity prediction of SARS-CoV-2 main protease inhibitors using recurrent neural network-based transfer learning. BMC Chem. 2021 Feb 2;15(1):8. doi: 10.1186/s13065-021-00737-2. PMID: 33531083; PMCID: PMC7852053.
- Saakre M, Mathew D, Ravisankar V. Perspectives on plant flavonoid quercetin-based drugs for novel SARS-CoV-2. Beni Suef Univ J Basic Appl Sci. 2021;10(1):21. doi: 10.1186/s43088-021-00107-w. Epub 2021 Mar 24. PMID: 33782651; PMCID: PMC7989718.
- Das S, Sarmah S, Lyndem S, Singha Roy A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn. 2021 Jun;39(9):3347-3357. doi: 10.1080/07391102.2020.1763201. Epub 2020 May 13. PMID: 32362245; PMCID: PMC7232884.
- M Khan MT, Ali A, Wang Q, Irfan M, Khan A, Zeb MT, Zhang YJ, Chinnasamy S, Wei DQ. Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2-a molecular dynamic study. J Biomol Struct Dyn. 2021 Jul;39(10):3627-3637. doi: 10.1080/07391102.2020.1769733. Epub 2020 Jun 1. PMID: 32410504; PMCID: PMC7284144.
- Joshi T, Sharma P, Joshi T, Pundir H, Mathpal S, Chandra S. Structure-based screening of novel lichen compounds against SARS Coronavirus main protease (Mpro) as potentials inhibitors of COVID-19. Mol Divers. 2021 Aug;25(3):1665-1677. doi: 10.1007/s11030-020-10118-x. Epub 2020 Jun 29. PMID: 32602074; PMCID: PMC7323881.
- Sk MF, Roy R, Jonniya NA, Poddar S, Kar P. Elucidating biophysical basis of binding of inhibitors to SARS-CoV-2 main protease by using molecular dynamics simulations and free energy calculations. J Biomol Struct Dyn. 2021 Jul;39(10):3649-3661. doi: 10.1080/07391102.2020.1768149. Epub 2020 Jun 1. PMID: 32396767; PMCID: PMC7284146.
- Bhardwaj VK, Singh R, Sharma J, Rajendran V, Purohit R, Kumar S. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J Biomol Struct Dyn. 2021 Jul;39(10):3449-3458. doi: 10.1080/07391102.2020.1766572. Epub 2020 May 20. PMID: 32397940; PMCID: PMC7256349.
- Qu H, Zheng Y, Wang Y, Li H, Liu X, Xiong X, Zhang L, Gu J, Yang G, Zhu Z, Zheng H, Ouyang Q. The potential effects of clinical antidiabetic agents on SARS-CoV-2. J Diabetes. 2021 Mar;13(3):243-252. doi: 10.1111/1753-0407.13135. Epub 2020 Dec 19. PMID: 33210826; PMCID: PMC7753367.
- Rabie AM. RETRACTED ARTICLE: Discovery of (E)-N-(4-cyanobenzylidene)-6-fluoro-3-hydroxypyrazine-2-carboxamide (cyanorona-20): the first potent and specific anti-COVID-19 drug. Chem Zvesti. 2021;75(9):4669-4685. doi: 10.1007/s11696-021-01640-9. Epub 2021 May 16. Retraction in: Chem Zvesti. 2022 Jun 4;:1. PMID: 34025012; PMCID: PMC8126404.
- Gao H, Sun X, Rao Y. PROTAC Technology: Opportunities and Challenges. ACS Med Chem Lett. 2020 Mar 12;11(3):237-240. doi: 10.1021/acsmedchemlett.9b00597. PMID: 32184950; PMCID: PMC7073876.
- Di Sarno V, Lauro G, Musella S, Ciaglia T, Vestuto V, Sala M, Scala MC, Smaldone G, Di Matteo F, Novi S, Tecce MF, Moltedo O, Bifulco G, Campiglia P, Gomez-Monterrey IM, Snoeck R, Andrei G, Ostacolo C, Bertamino A. Identification of a dual acting SARS-CoV-2 proteases inhibitor through in silico design and step-by-step biological characterization. Eur J Med Chem. 2021 Dec 15;226:113863. doi: 10.1016/j.ejmech.2021.113863. Epub 2021 Sep 22. PMID: 34571172; PMCID: PMC8457654.
- Andrzejczyk, J, Jovic, K, Brown, LM, Pascetta, VG, Varga, K, Vashisth, H(2022). Molecular interactions and inhibition of the SARS-CoV-2 main protease by a thiazolidinedione derivative. Proteins; 1- 12.https:// doi/10.1002/prot.26385
- Mediouni S, Mou H, Otsuka Y, Jablonski JA, Adcock RS, Batra L, Chung DH, Rood C, de Vera IMS, Rahaim R Jr, Ullah S, Yu X, Getmanenko YA, Kennedy NM, Wang C, Nguyen TT, Hull M, Chen E, Bannister TD, Baillargeon P, Scampavia L, Farzan M, Valente ST, Spicer TP. Identification of potent small molecule inhibitors of SARS-CoV-2 entry. SLAS Discov. 2022 Jan;27(1):8-19. doi: 10.1016/j.slasd.2021.10.012. Epub 2021 Oct 23. PMID: 35058179; PMCID: PMC8577999.
- Singh P, Chauhan SS, Pandit S, Sinha M, Gupta S, Gupta A, Parthasarathi R. The dual role of phytochemicals on SARS-CoV-2 inhibition by targeting host and viral proteins. J Tradit Complement Med. 2022 Jan;12(1):90-99. doi: 10.1016/j.jtcme.2021.09.001. Epub 2021 Sep 8. PMID: 34513611; PMCID: PMC8424525.
- Tang B, He F, Liu D, He F, Wu T, Fang M, Niu Z, Wu Z, Xu D. AI-Aided Design of Novel Targeted Covalent Inhibitors against SARS-CoV-2. Biomolecules. 2022; 12(6):746. https://doi.org/10.3390/biom12060746
- Benítez-Cardoza CG, Vique-Sánchez JL. Potential inhibitors of the interaction between ACE2 and SARS-CoV-2 (RBD), to develop a drug. Life Sci. 2020 Sep 1;256:117970. doi: 10.1016/j.lfs.2020.117970. Epub 2020 Jun 15. PMID: 32553928; PMCID: PMC7294299.
- Khaerunnisa S, Kurniawan H, Awaluddin R, Suhartati S, Soetjipto S. Potential Inhibitor of COVID- 19 Main Protease (Mpro) from Several Medicinal Plant Compounds by Molecular Docking Study. Preprints. 2020. doi: 10.20944/preprints202003.0226.v1
- Dallakyan S, Olson AJ. Small-Molecule Library Screening by Docking with PyRx. In: Hempel, J., Williams, ., Hong, C. (eds) Chemical Biology. Methods in Molecular Biology. 2015; 1263. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-2269-7_19
- Roymans D, De Bondt HL, Arnoult E, Geluykens P, Gevers T, Van Ginderen M, Verheyen N. Binding of a potent small-molecule inhibitor of six-helix bundle formation require interactions with both heptad-repeats of the RSV fusion protein. PNAS https://doi.org/10.1073/pnas.091010810
- Al-Zrkani MK, Abdulkareem RA, Al-Fahad D, Al Shouber M, Nasr AMS, Al-Khdhairawi A. Elucidating novel antibacterial compounds from the NPASS database against the FimH lectin domain for the treatment of urinary tract infections: an in-silico study. J Biomol Struct Dyn. 2022 Apr 9:1-12. doi: 10.1080/07391102.2022.2059009. Epub ahead of print. PMID: 35403563.
- Rauf A, Raza M, Humayun Khan M, Hemeg HA, Al-Awthan YS, Bahattab O, Bawazeer S, Naz S, Basoglu F, Saleem M, Khan M, Seyyedamirhossein H, Mubarak MS, Erdogan Orhan I. in vitro and in silico studies on clinically important enzymes inhibitory activities of flavonoids isolated from Euphorbia pulcherrima. Ann Med. 2022 Jan 27;54(1):495-506. doi: 10.1080/07853890.2022.2033826. PMID: 35112936; PMCID: PMC8820783.
- Goodsell DS, Morris GM, Olson AJ. Automated docking of flexible ligands: applications of AutoDock. J Mol Recognit. 1996 Jan-Feb;9(1):1-5. doi: 10.1002/(sici)1099-1352(199601)9:1<1::aid-jmr241>3.0.co;2-6. PMID: 8723313.
- Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000 Jul-Aug;44(1):235-49. doi: 10.1016/s1056-8719(00)00107-6. PMID: 11274893.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017 Mar 3;7:42717. doi: 10.1038/srep42717. PMID: 28256516; PMCID: PMC5335600.
- Mustafa M, Mostafa YA. Antimicrobial Pyridazines: Synthesis, Characterization, Cytotoxicity, Substrate Promiscuity, and Molecular Docking. Chem Biodivers. 2020 Jun;17(6):e2000100. doi: 10.1002/cbdv.202000100. Epub 2020 May 8. PMID: 32239712.
- Khadse SC, Amnerkar ND, Dave MU. Quinazolin-4-one derivatives lacking toxicity-producing attributes as glucokinase activators: design, synthesis, molecular docking, and in-silico ADMET prediction. Fut J Pharm Sci; 2019; 5: 11. https://doi.org/10.1186/s43094-019-0012-y