
More Information
Submitted: December 04, 2023 | Approved: December 20, 2023 | Published: December 21, 2023
How to cite this article: Fathima S, Kishan V. Studies on the Influence of Charge Inducer and it’s Combination with P-gp Inhibitor to Improve the Oral Bioavailability of Nimodipine via Submicron Lipid Emulsions. Arch Pharm Pharma Sci. 2023; 7: 074-082.
DOI: 10.29328/journal.apps.1001046
Copyright License: © 2023 Fathima S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and repro-duction in any medium, provided the original work is properly cited.
Keywords: Nimodipine; Submicron emulsion; Oral bioavailability; Pharmacokinetic; P-gp inhibitor

Studies on the Influence of Charge Inducer and it’s Combination with P-gp Inhibitor to Improve the Oral Bioavailability of Nimodipine via Submicron Lipid Emulsions
Sara Fathima and Veerabrahma Kishan*
Department of Pharmaceutics, Laboratory of Nanotechnology, University College of Pharmaceutical Sciences, Kakatiya University, Warangal, Telangana -506009, India
*Address for Correspondence: Veerabrahma Kishan, Department of Pharmaceutics, Laboratory of Nanotechnology, University College of Pharmaceutical Sciences, Kakatiya University, Warangal, Telangana -506009, India, Email: [email protected]
Background: Nimodipine (NM), is a dihydropyridine calcium channel blocker with poor oral bioavailability (BA) of about 13% due to first-pass metabolism and P-gp efflux.
Objective: The present work aimed to study the influence of the charge inducer and its combination with P-gp inhibitor to improve the oral bioavailability of NM by developing a suitable delivery system of Submicron Lipid Emulsion (SME).
Methods: Five SME formulations of NM were prepared by homogenization followed by ultrasonication. Prepared SMEs were characterized for particle size, PDI, Zeta Potential (ZP), Entrapment Efficiency (EE), and drug content. In vitro, release studies were performed in 0.1N HCl and pH 6.8 phosphate buffer by open tube method. The physical stability of all NM–SMEs was tested by the individual effects of centrifugation, dilution (desorption stress), and storage. Bioavailability studies were conducted on male Wistar rats after oral administration of NM suspension and F1 to F5 SME formulations.
Results and conclusion: Five NM- SMEs were developed with a mean size ranging from 93 - 137 nm, Zeta potential of – 26 ± 1 mV (negatively charged), +45.8 to +46.3 mV (positively charged), and PDI of 0.15 - 0.25. The in vitro release studies showed that relatively more cumulative percentage release of NM – SMEs in 0.1N HCl than in pH phosphate buffer during 24 hours. The physical stability of NM–SMEs indicated that they were stable to the effects of centrifugation, dilution, and storage. Pharmacokinetic (PK) studies showed that the oral bioavailability of NM in F4 SME was significantly higher than that of all other formulations. Taken together, the results indicated the development of a stable lipid-based carrier, F4 SME to improve the oral bioavailability of this drug by minimizing first-pass metabolism due to lymphatic transport, reducing the efflux by P-gp inhibition, and further, by increased uptake of the positively charged F4 SME globules by enterocytes. Future: The research study findings increase the possibility of developing NM F4 SME by the pharmaceutical industry for the patient’s benefit.
Drugs through the oral route achieve very high compliance, as it is patient-friendly and easy to swallow. However, the oral route of drug delivery offers challenges for drugs having poor solubility, and chemical instability in the gastrointestinal tract. In general, about 40% of newly developed drugs are not suitable for oral absorption [1]. Hence, these are potential candidates for improvement of Bioavailability (BA). Here, lipid-based delivery systems like Lipid Nanoemulsions (LNEs), Solid Lipid Nanoparticles (SLNs), etc., play an important role. Oral drug delivery has taken a new dimension with the increasing application of lipids as a carrier for the delivery of poorly water-soluble, lipophilic drugs [2]. Submicron (o/w) lipid emulsions (SME) are potential drug carriers for lipophilic and amphiphilic drugs and possess many favorable properties: they are biocompatible, biodegradable, stable, and easy to prepare and handle. The basic structure of SME has a neutral lipid core (i.e., triglyceride), stabilized by a monolayer of amphiphilic lipid (i.e., phospholipids), and dispersed in aqueous phase. Such emulsions can solubilize a considerable amount of lipophilic drugs in the core /and amphiphilic ones in the surface monolayer. Submicron lipid emulsions are prepared by emulsifying biocompatible oil (or) triglyceride-containing lipid-soluble drugs using homogenizer and/or ultrasonicator, lecithin (phospholipids) being added as an emulsifying agent [3]. The size of globules ranged from 200 nm – 600 nm.
In general, a negative charge is present on the surface of globules of SME and that of enterocytes. For better uptake of SME globules by enterocytes, charge–charge interactions are important. To improve the uptake by enterocytes, and subsequent oral bioavailability of the drug through SMEs, a positive charge on the surface of SME globules plays an important role. The positive charges can be obtained by adding a positive charge inducer like stearylamine, at the time of preparation [4]. This addition would induce positive charges on the surfaces of SME globules. Interaction of enterocytes and positively charged oil globules of SME contribute to increased uptake in the gastrointestinal tract and thus increased oral bioavailability [4]. Further, the high lipophilic nature of the drug favors lymphatic transport. This reduces the first-pass effect. In cases, where the P-gp efflux transporter plays a role in reducing the bioavailability by pumping the drug out of cells, e. g., enterocytes. This efflux could be reduced by adding a P-gp inhibitor in the formulation, an example like tween 80, which acted as co- surfactant [5]. Tween 80 inclusion contributed to increased absorption of Digoxin, a P-gp substrate [6]. This is a promising approach to increase the bioavailability of formulations [7].
Nimodipine (NM), is a dihydropyridine calcium antagonist with therapeutic indications for cerebrovascular spasm, stroke, and migraine. NM is a BCS class II drug with low solubility and high permeability [8]. However, the clinical efficacy of NM is strongly limited by its poor water solubility (2.30 µg/ml) and low oral bioavailability (~13%) [9]. It has a logP 3.41 [10]. Apart from the poor water solubility, extensive first-pass metabolism by Cytochrome P450-3A4 (CYP3A4) iso-enzymes and P-glycoprotein (P-gp) mediated efflux is also responsible for the low oral bioavailability of NM [9]. Many trials were made to improve the dissolution or bioavailability of nimodipine by developing a variety of delivery systems. Dissolution medium improves the rate of dissolution [11]. In this context, nimodipine was developed as solid dispersions [8], SEDDS [9], liquid proliposomes [12], nanostructured lipid carriers [13], SLNs [10], microemulsion [14,15] and lipid microspheres [16]. To overcome hepatic first-pass metabolism, and insolubility of drug and to enhance oral bioavailability, lipid-based drug delivery systems like SLNs, and SMEs were studied [17-19]. Nanoemulsions and nanonization of poorly soluble drugs improve the bioavailabilty [20,21]. Nano emulsions can be designed for parenteral use [22-24]. Submicron emulsions (SME) of NM have not been reported till now. SLNs of NM [10] were focused on improving intestinal lymphatic transport. In a similar manner, the submicron lipid emulsion of this study is also expected to improve lymphatic transport and thus contribute to reduced first pass effect of NM. Further, the role of cationic charge inducer, stearyl amine, and P-gp efflux inhibitor, tween 80, and its combination for improvement of BA is not yet reported.
In the present study, SMEs were prepared individually, with a positive charge inducer and a P-gp inhibitor. Further, a combination of P-gp inhibitor and positive charge inducer was used in SME as a delivery strategy to improve the oral bioavailability of nimodipine. It was expected to increase the globule uptake by enterocytes, reducing the first-pass metabolism by increasing lymphatic transport and inhibiting P-gp mediated efflux of drug and further, a combinational effect of P-gp inhibitor and positive charge inducer by inclusion in SME delivery system.
Materials
Nimodipine was obtained as a gift sample from Dr Reddy´s Laboratories, Hyderabad, India. Soybean oil and olive oil were purchased from Sigma-Aldrich Chemicals, Mumbai, India. Egg Lecithin E-80 was a gift sample from Lipoid, Germany. Methanol, acetonitrile, and dichloromethane were of HPLC grade and were purchased from Merck Ltd. (Mumbai, India). Centrisart membrane filters (molecular weight cut off 20,000) were purchased from Sartorius, Goettingen, Germany.
Oil solubility studies
The solubility of NM was studied in three different oils i.e., olive oil, soybean oil, and sunflower oil. Accurately weighed 25 mg of drug was added to 2ml of oil, taken in a vial. The vial was tightly closed and kept for shaking on a gyratory shaker (GFL, Germany) for 48 hours at 180 rpm.
Solubility of the drug was also conducted using egg lecithin. Accurately weighed amounts of 25 mg of the drug and 120 mg of phosphatidy lcholine EPC-80 were added to the 2 ml of oil in a vial and subjected to mild heating until egg lecithin was molten.
After shaking for 48 hours, contents were centrifuged (Biofuge primo R, Heraeus) for 20 min. The obtained supernatant was again centrifuged for another 15 minutes. From this 0.1mL of supernatant was collected and diluted to 1mL with CHCl3: Methanol (1:1). The final volume was made up to 5 mL with mobile phase 68:32 (Acetonitrile: Water). The clear solution obtained was injected into HPLC. The solubility of the drug was determined by using the reported HPLC method and a developed standard graph [25].
HPLC method was developed based on a validated method [25] to estimate oil solubility, total drug content, and entrapment efficiency of the system. An accurately weighed amount of 10 mg of nimodipine was dissolved in 10 mL of methanol (stock solution-I). About 0.1 mL was taken from stock I and diluted to 1 mL with mobile phase (stock solution II). Different dilutions were made with the mobile phase to obtain 0.025, 0.05, 0.1, 0.25, 0.5, 1, 2.5, 5, and 10 µg/mL. The solutions were spiked onto the HPLC column. The standard graph was plotted based on peak area (peak area Vs concentration) from the obtained chromatogram [25].
Chromatographic conditions
Mobile phase: Acetonitrile: Water (68:32)
Flow rate: 1 ml/min
Column: C-18 (250 mm X 4.6 mm i.d., 5 µm)
Column temperature: 25 °C
UV- detection at: 240nm
Detector sensitivity: 0.0005 AUFS
Injection volume: 20 µl
Retention time: 7.2 min
Preparation of SMEs
Formation of stable emulsions is needed [26,27]. In all, five NM SME formulations were prepared by homogenization followed by ultrasonication method [4] using ingredients as mentioned in Table 1. Positive charge and negative charges were obtained on SMEs by adding oleic acid as a negative charge inducer and stearylamine as positive charge inducer (F1, F2). Tween 80 was added to inhibit P-gp efflux in two SMEs (F3, F4). Further in F3 and F4, oleic acid and stearylamine were added for inducing negative and positive charges respectively. F5 SME as control plain emulsion, without any added charge inducer and P-gp inhibitor. Drug (NM), egg lecithin, cholesterol, α- tocopherol acetate, and oleic acid were dissolved in soybean oil, heated to 70 °C in a water bath, and stirred until the system was clear. Glycerol was dissolved in a sufficient amount of double distilled water and the aqueous phase was also heated to 70 °C and then it was added to the oil phase at the same temperature.
| Table 1: Compositions of prepared NM SMEs (F1 - F5) and suspension (F6). | ||||||
| Formulation ingredients | Formulation code | |||||
| F1 | F2 | F3 | F4 | F5 | F6 | |
| Organic Phase | ||||||
| NM (mg) | 7.5 | 7.5 | 7.5 | 7.5 | 7.5 | - |
| Soybean Oil ( g) | 1 | 1 | 1 | 1 | 1 | - |
| Egg lecithin, EPC - 80 (mg) | 120 | 120 | 120 | 120 | 120 | - |
| Cholesterol (mg) | 30 | 30 | 30 | 30 | 30 | - |
| Oleic acid (mg) | 25 | - | 25 | - | - | - |
| Stearyl amine (mg) | - | 30 | - | 30 | - | - |
| α- tocopherol acetate (mg) | 20 | 20 | 20 | 20 | 20 | - |
| Aqueous Phase | ||||||
| NM (mg) | - | - | - | - | - | 7.5 |
| Glycerol ( mg) | 225 | 225 | 225 | 225 | 225 | - |
| Tween 80 ( 1%) (mg) | - | - | 100 | 100 | - | - |
| Sodium carboxy methyl cellulose (mg) | - | - | - | - | - | 50 |
| Double distilled water (mL) | 10 | 10 | 10 | 10 | 10 | 10 |
A coarse emulsion (10 mL) was prepared by homogeni-zation (Heidolph homogenizer DLX 900) for 3 min at 15000 rpm. The homogenized emulsion was further subjected to ultrasonication by Sonics’ ultrasonicator (USA) for 20 min with a 12 T probe.
Preparation of nimodipine suspension
About 50 mg sodium carboxy methyl cellulose (suspending agent) was taken in a mortar and triturated for 3 min, then 7.5 mg of nimodipine was added to it and triturated for 3 min. To it, 10 mL of double distilled water was added and again triturated for 5 min to form nimodipine suspension (F6).
Characterization of SMEs
Determination of particle size, PDI, and Zeta potential of SMEs: The prepared SME preparations were diluted in a 1:50 ratio with double distilled water and size was measured at 90° angles by Malvern Zeta sizer (Nano ZS 90). The average particle size, polydispersity index, and zeta potential were obtained directly from the instrument [4].
Physical stability of submicron lipid emulsions: Here, the centrifugal and dilution stresses were applied to the prepared SMEs.
Effect of centrifugation: The effect of centrifugation can be determined by creaming volume percentage. About 1 mL of submicron lipid emulsions were taken into centrifuge tubes, and the weights were balanced and kept diagonally in a centrifuge (Biofuge Primo R, Heraeus). The emulsions were centrifuged at 15,000 rpm (1529 × g) for 10 minutes, to assess the stability of emulsions under centrifugation. The creaming volume percentage for each emulsion was calculated by using the formula [28] and then compared.
Where C = Creaming volume percentage, Vt = Total volume of sample, Vs = Volume of the lower phase layer.
Effect of dilution (desorption stress)
The prepared submicron lipid emulsions were subjected to dilutions (1:50, 1:100, 1:500, and 1:1000) with double distilled water. Then the effect of dilution on the size of globules, PDI, zeta potential, and stability was assessed.
Short-period stability of SMEs under storage: The short time stability of prepared NM-SMEs was studied by storing them at room temperature, and at 4 °C for two months and observed for size, PDI, and zeta potential, and further visually observed for any breakage of emulsion.
Determination of entrapment efficiency (EE) and total drug content by HPLC: Entrapment Efficiency (EE) was determined by measuring the concentration of free drug (unentrapped) in aqueous medium. The aqueous medium was separated by ultra-filtration using centrisart tubes (Sartorius, Goettingen, Germany), which consisted of a filter membrane (M. wt. cut off 20,000 Da) at the base of the sample recovery chamber. About 2.5 mL of the formulation was placed in the outer chamber and a sample recovery chamber was placed over the sample and centrifuged at 3500 rpm for 15 mins. The SME along with the encapsulated drug remained in the outer chamber and the aqueous phase moved into the sample recovery chamber through a filter membrane. The amount of nimodipine in the aqueous phase was estimated by using a reported validated HPLC method [25]. The column and other conditions were described in the above text.
Determination of total drug content
About 100 µL of the formulation was dissolved in 1 mL with methanol (HPLC Grade) and then further dilutions were made with mobile phase. The diluted samples were estimated by the HLPC method and nimodipine content was estimated.
In vitro drug release studies: The dialysis method was used to perform in vitro release studies. Dialysis membrane (Himedia, Mumbai) having a pore size of 2.4 nm and molecular weight cut off between 12000 – 14000 was used for the release studies. The dialysis membrane was soaked overnight in double distilled water prior to the release studies. Hydrochloric acid (0.1N) and phosphate buffer pH 6.8 were used as release media. The experimental unit had donor and receptor compartments. The donor compartment consisted of a boiling tube which was cut open at one end and tied with a dialysis membrane at the other end into which 1 mL of SME dispersion was taken for release study. The receptor compartment consisted of a 250 mL beaker which was filled with 100 mL of a release medium (70: 30) containing 70 mL (0.1 N HCl and 6.8 pH phosphate buffer) and 30 mL of ethanol and temperature was maintained at 37 ± 0.5 °C. At 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24-hour time points, 1 mL samples were withdrawn from the receiver compartment and replenished with the same volume of release medium. The collected samples were suitably diluted and analyzed by UV-visible spectrophotometer (SL-150, ELICO) at 240 nm.
Pharmacokinetic study of SMEs: The study objective was to find out the individual oral bioavailability of five prepared nimodipine SMEs (F1- F5) and compare it with that of a nimodipine suspension (F6). The study design was one-time, single dose, oral administration, six groups of rats (n = 6). The study was conducted with prior approval of “Institutional Animal Ethical Committee” (File no. IAEC/02/UCPSc/KU/2012). The male albino Wistar rats weighing 180 gm - 220 gm were divided into six groups (6 animals each, 6*6 = 36 rats) and were randomly orally administered at a single dose of 5 mg/kg body weight for each nimodipine submicron lipid emulsion formulations (F1 - F5) and F6 control suspension. Overnight fasted animals with adequate water were used. At pre-determined time intervals (0.5, 1, 2, 4, 6, 8, 10, 12, and 24 hrs), the blood samples of 0.3 mL – 0.4 mL were collected by puncturing the retro-orbital venous plexus. The blood was allowed to clot, then centrifuged at 4000 rpm for 15 mins and serum was collected. The serum samples were stored at -20 °C until analysis.
Extraction procedure
In general, liquid-liquid extraction method is used in this category of drugs [29]. To 100 µL of serum, 25 µL each of methanol and 5 µg/mL solution of felodipine (Internal standard) were added and vortexed for 2 min. Then about 100 µL of 1 N sodium hydroxide solution (1N NaOH) was added and vortexed for 3 min. Then 750 µL of dichloromethane was added and vortexed for 5 min followed by centrifugation at 5,000 rpm for 15 min. The organic phase was separated and evaporated under reduced pressure in a vacuum oven (Toshniwal, India). The residue was reconstituted with 50 µL of mobile phase and 20 µL of the reconstituted sample was spiked onto the HPLC (LC 10AT, Shimazdu, Japan) with a UV-detector (SPD 10A, Shimazdu, Japan).
HPLC conditions: Chromatographic conditions
Mobile phase: Acetonitrile: water (70:30)
Flow rate: 1 mL/min
Column: Lichrosphere C18 (250 mm X 4.6 mm i.e., 5 µm particle size)
Column temperature: 25 °C
Injection volume: 20 µL
UV – detection: 237 nm
Detector sensitivity: 0.0005 AUFS
Retention time: 6.79 min for drug and 8.78 min for felodipine(internal standard).
Standard graph of NM: The peak areas ratio of NM to that of internal standard (felodipine) was used for the quantification of NM in serum samples. The calibration curve was linear in the concentration range of 0.01 - 4 µg/mL. The regression equation is y = mx + c, where y represents the peak ratio of NM to I.S, x represents the concentrations of NM, m is the slope of the curve and c is the intercept. The calibration graph was obtained in rat serum with y = 0.6394x + 0. 0492 (r 2 = 0.997).
Calculation of pharmacokinetic parameters: The concentrations of nimodipine in rat serum samples were obtained from the standard graph prepared. The pharmacokinetic parameters Cmax, Tmax, AUC0-24, MRT, and t1/2 were calculated by Kinetica software, 2000.
Statistical analysis: The statistical comparison of data was done using an unpaired t-test by GraphPad Prism software (version 5.0, 2007), and significance was calculated at a p - value of 0.001.
Oil solubility studies
Three different oils were checked for solubility and results are shown in Table 2. Among them, soybean oil showed maximum solubility of 4.341 ± 0.008 mg/ mL. To further enhance the solubility of the drug, phosphatidyl choline (EPC-80) was added to oils. Accordingly, the soybean oil and PC combination increased the NM drug solubility to 7.622 ± 0.022 mg/mL. The chosen delivery system SME contained a PC as a co-surfactant. Hence, it was expected to increase the loading of NM drug in this delivery system.
| Table 2: Oil solubility studies of NM in different oils. | |
| Oils | Solubility (mg/mL) |
| Olive oil | 3.621 ± 0.008 |
| Sunflower oil | 2.644 ± 0.002 |
| Soybean oil | 4.341 ± 0.008 |
| Olive oil + phosphatidylcholine | 5.140 ± 0.043 |
| Sunflower oil + phosphatidylcholine | 3.160 ± 0.002 |
| Soybean oil + phosphatidylcholine | 7.622 ± 0.022 |
Preparation and characterization of SMEs formulation: The submicron lipid emulsions were prepared by homogenization followed by ultra-sonication for different formulate.ons i.e., F1 - F5. All the prepared samples were analyzed to determine their particle size, zeta potential, and PDI values. The results are presented in Table 3. The particle size of all formulations ranged from 93.1 ± 4.55 to 136.8 ± 11.76. PDI from 0.15 ± 0.004 to 0.25 ± 0.036 and zeta potential negatively charged from -20.1 ± 2.15 to – 26.5 ± 1.02 and positively charged + 45.8 ± 3.11 to + 46.3 ± 1.89. Here, in this study, all the formulations showed lower particle sizes, PDI was also less than 0.3, which was within the permissible limits and zeta potentials were also moderate and satisfactory.
Among them F1 (negatively charged) formulation exhibited the lowest globule size. F5 (control) showed the highest particle size.
The entrapment efficiency and drug content were determined by HPLC for all formulations and the results are shown in Table 3. All formulations showed entrapment efficiency ranging from 99.50 ± 0.421 to 99.90 ± 0.008. All formulations showed higher values and were not significantly different. The total drug content of the SME formulation was determined and found in the range to be 7.33 ± 0.13 mg to 7.46 ± 0.30 mg.
| Table 3: Physical characters of prepared SMEs. | |||||
| Formulation code | Size (nm) | PDI | Zp (mV) | Total drug content (mg) |
Entrapment efficiency (%) |
| F1 | 93.1 ± 4.55 | 0.17 ± 0.027 | -26.5 ± 1.02 | 7.35 ± 0.20 | 99.50 ± 0.421 |
| F2 | 128.7 ± 6.58 | 0.23 ± 0.025 | +45.8 ± 3.11 | 7.41± 0.170 | 99.85 ± 0.005 |
| F3 | 125.2 ± 13.28 | 0.20 ± 0.154 | -25.0 ± 1.74 | 7.46 ± 0.30 | 99.95 ± 0.012 |
| F4 | 103.7 ± 9.39 | 0.25 ± 0.036 | +46.3 ± 1.89 | 7.33 ± 0.13 | 99.91 ± 0.002 |
| F5 | 136.8 ± 11.76 | 0.15 ± 0.004 | -20.1 ± 2.15 | 7.39 ± 0.42 | 99.90 ± 0.008 |
In vitro release studies
In vitro, release studies were performed for all formulations in 0.1 N HCl and pH 6.8 phosphate buffer media for 24 hr. Sink condition was ensured [30] in this study. At pH 1.2, the cumulative percent drug released from F6 (control suspension) was 87.73 and from other formulations F1, F2, F3, F4, and F5, they were 76.40, 64.5, 75.93, 67.27, and 79.52 respectively (Figure 1). At pH 6.8, the cumulative percent release of drug from formulation F6 was 83.41, whereas for formulations F1, F2, F3, F4, and F5 were 81.9, 67.74, 78.43, 65.9, and 75.89 respectively (Figure 2).
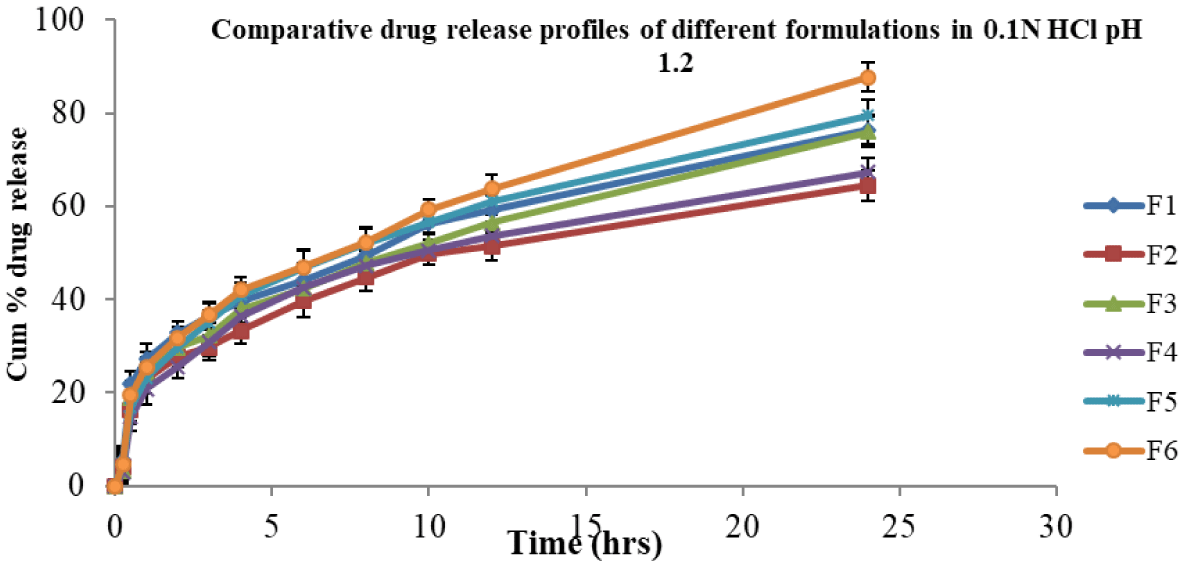
Figure 1: Cumulative % drug release from nimodipine SMEs in 0.1N HCl.
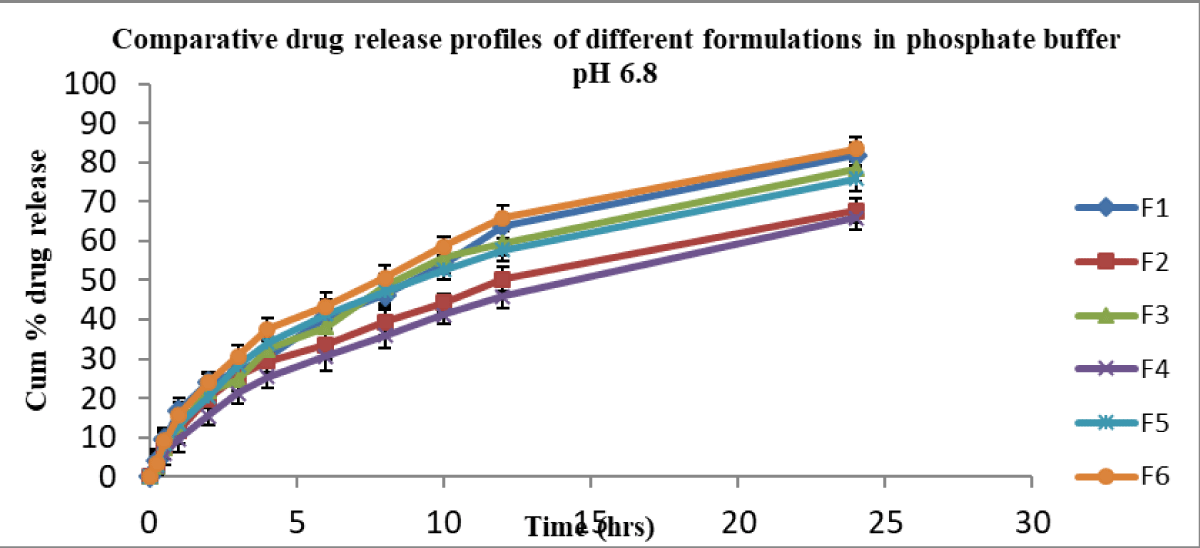
Figure 2: Cumulative % drug release from nimodipine SMEs in pH 6.8 phosphate buffer.
The formulations (F1 – F5) released in 0.1 N HCl below 80% in 24 hrs and were not significantly different at various time points. Further, F1 – F5 formulations released in pH 6.8 phosphate buffer below 82% in 24 hrs. This clearly indicated that irrespective of dissolution medium pH 1.2 or pH 6.8, the release profiles were not significantly different. The suspension showed a slight improvement in release at 24 hrs irrespective of medium pH condition over SMEs.
In the above two cases, the release of drug from formula-tions (F1 – F5) was almost similar and less than 82%. None of the formulations released above 90% in 24 hrs. Formulations F1, F2, F3 and F4 (charged emulsions) contained charge inducers (positive or negative) along with cholesterol which would rigidise the interface, in turn reduce the release of the drug [4]. When compared, the plain emulsion F5 (control) of this study, without any added charge inducer and co-surfactant, could show almost similar release profile (Figure 1). In contrast, the release of drug from negatively charged (F1, F3) was slightly more than that of positively charged SMEs (F2, F4). Here, the average globule size of the control SME (F5) was bigger than that of other formulations (F1 ̶ F4). Both the rigidity of the surfactant (F1 to F4) and the big size (F5) of the globule contributed similarly to the cumulative % drug release.
Overall, the cumulative % release of nimodipine from all the formulations was not the same, irrespective in 0.1 N HCl or in pH 6.8 phosphate buffer medium. About 50% of the drug was released in 8 hrs - 10 hrs in 0.1 N HCl medium, whereas it was 10 hrs - 12 hrs in pH 6.8 phosphate buffer for all formulations.
Physical stability studies
Dilution and centrifugal effects: The stability of nimodipine SMEs was determined by studying the effects of centrifugation and desorption. The effect of centrifugation was assessed by % creaming volumes and was determined for all formulations and all of them showed higher % creaming volumes which indicated good stability and the values are given in Table 4. There were no differences noticed among all the SMEs. Further dilution effect was determined by diluting all formulations in the ratio of 1:50, 1:100, 1:500, 1:1000.
| Table 4: Effect of centrifugation on the stability of SMEs: Creaming volume percentage of prepared formulations. | |
| Formulation code | % Creaming volume v/v |
| F1 | 98.43 ± 0.64 |
| F2 | 97.31 ± 0.52 |
| F3 | 99.12 ± 0.21 |
| F4 | 99.62 ± 0.26 |
| F5 | 98.55 ± 0.41 |
Changes noticed upon dilution are given in Table 5. There was no significant difference found in size, PDI, and zeta potential of the formulations during the dilution up to 500 times. However, at 1000 dilution times slight changes in size were noticed. Apparently, it indicated that all the SMEs were stable.
| Table 5: Effect of dilution (desorption stress) on globule size and zeta potential. | ||||
| Formulation | Dilution factor | Size ( nm) | PDI | Zp (mV) |
| F1 | 1:50 | 92.5 ± 2.12 | 0.158 ± 0.03 | -26.5 ± 4.60 |
| 1:100 | 99.7 ± 3.22 | 0.165 ± 0.01 | -24.3 ± 3.63 | |
| 1:500 | 106.1 ± 5.91 | 0.178 ± 0.02 | -25.2 ± 2.88 | |
| 1:1000 | 120.5 ± 4.63 | 0.182 ± 0.04 | -26.1 ± 2.77 | |
| F2 | 1:50 | 125.6 ± 2.32 | 0.227 ± 0.01 | +46.3 ± 3.91 |
| 1:100 | 132.6 ± 3.45 | 0.234 ± 0.03 | +46.3 ± 2.65 | |
| 1:500 | 141.2 ± 1.24 | 0.238 ± 0.02 | +47.2 ± 4.73 | |
| 1:1000 | 153.1 ± 6.13 | 0.246 ± 0.04 | +45.6 ± 1.27 | |
| F3 | 1:50 | 121.1 ± 3.42 | 0.209 ± 0.12 | -27.4 ± 5.90 |
| 1:100 | 126.0 ± 2.33 | 0.211 ± 0.04 | -28.2 ± 2.13 | |
| 1:500 | 130.4 ± 4.21 | 0.215 ± 0.02 | -26.1 ± 1.34 | |
| 1:1000 | 141.7 ± 3.16 | 0.225 ± 0.06 | -27.7 ± 3.47 | |
| F4 | 1:50 | 106.7 ± 5.60 | 0.231 ± 0.01 | +45.2 ± 5.05 |
| 1:100 | 110.8 ± 4.91 | 0.242 ± 0.02 | +46.4 ± 1.81 | |
| 1:500 | 115.9 ± 6.31 | 0.245 ± 0.03 | +45.4 ± 2.24 | |
| 1:1000 | 126.7 ± 2.70 | 0.252 ± 0.04 | +44.6 ± 3.28 | |
| F5 | 1:50 | 137.8 ± 3.45 | 0.154 ± 0.04 | -22.1 ± 3.26 |
| 1:100 | 141.1 ± 2.65 | 0.169 ± 0.01 | -20.2 ± 1.74 | |
| 1:500 | 148.0 ± 5.88 | 0.178 ± 0.03 | -21.4 ± 2.21 | |
| 1:1000 | 159.8 ± 1.85 | 0.182 ± 0.04 | -22.3 ± 3.17 | |
Physical stability of SME under storage: Stability studies were conducted for all formulations at room temperature and refrigerated temperature for 2 months and the size, PDI, and zeta potential are given in Table 6. No significant changes were noticed in size, PDI, and zeta potential values, which indicated the stability during storage at room temperature and 4 °C for a two-month period.
| Table 6: Physical characters of SMEs during storage at RT and at 4 ○C. | |||||||||||||||
| At 25°C |
Size (nm) | PDI | Zeta potential (mV) | ||||||||||||
| Day | F1 | F2 | F3 | F4 | F5 | F1 | F2 | F3 | F4 | F5 | F1 | F2 | F3 | F4 | F5 |
| 0 | 95.4±5.6 | 132.3±5.7 | 129.4±6.2 | 105.1±5.6 | 135.1±6.8 | 0.12±0.05 | 0.21±0.06 | 0.18±0.01 | 0.23±0.02 | 0.17±0.02 | -26.2±1.1 | +45.3±2.5 | -25.6±6.2 | +46.5±5.1 | -20.2±2.3 |
| 15 | 100.1±4.7 | 135.2±4.9 | 132.2±4.7 | 109.4±4.8 | 139.3±7.2 | 0.13±0.09 | 0.22±0.03 | 0.20±0.03 | 0.25±0.06 | 0.20±0.01 | -27.3±3.2 | +44.1±2.6 | -26.2±5.1 | +47.7±2.2 | -21.5±3.4 |
| 30 | 103.5±8.0 | 139.5±8.3 | 136.1±3.6 | 112.6±2.6 | 141.2±7.5 | 0.14±0.03 | 0.22±0.05 | 0.21±0.04 | 0.24±0.01 | 0.21±0.07 | -25±1.4 | +47.5±1.4 | -27.3±2.7 | +46.8±3.6 | -22.3±6.4 |
| 45 | 107.3±5.5 | 142.7±8.1 | 140.2±2.6 | 116.8±8.5 | 144.6±6.9 | 0.14±0.02 | 0.23±0.01 | 0.22±0.03 | 0.26±0.04 | 0.23±0.05 | -26.3±1.5 | +45.3±3.5 | -25.5±3.1 | +45.3±6.5 | -20.4±3.9 |
| 60 | 116.7±7.3 | 149.0±6.7 | 146.0±4.8 | 122.2±5.7 | 151.2±2.4 | 0.15±0.01 | 0.24±0.06 | 0.24±0.08 | 0.27±0.03 | 0.26±0.04 | -24.5±3.2 | +44.6±5.3 | -27.8±3.7 | +44.3±1.6 | -19.8±5.4 |
| At 4°C |
Size (nm) | PDI | Zeta potential (mV) | ||||||||||||
| Day | F1 | F2 | F3 | F4 | F5 | F1 | F2 | F3 | F4 | F5 | F1 | F2 | F3 | F4 | F5 |
| 0 | 95.4±5.8 | 130.4±3.2 | 130.2±3.1 | 103.9±2.1 | 136.1±7.1 | 0.12±0.04 | 0.19±0.04 | 0.17±0.01 | 0.21±0.05 | 0.19±0.03 | -25.4±3.2 | +46.5±2.5 | -26.2±1.4 | +44.2±1.7 | -21.5±4.2 |
| 15 | 102.1±4.0 | 134.5±2.3 | 132.4±5.2 | 110.9±3.3 | 140.9±4.2 | 0.14±0.07 | 0.21±0.06 | 0.19±0.03 | 0.23±0.01 | 0.21±0.06 | -24.7±2.0 | +48.3±1.7 | -27.7±4.2 | +43.4±4.2 | -22.3±3.3 |
| 30 | 105.7±5.1 | 136.7±5.6 | 135.8±4.4 | 112.4±1.7 | 144.0±5.6 | 0.13±0.09 | 0.22±0.05 | 0.20±0.04 | 0.24±0.02 | 0.22±0.02 | -27.2±5.3 | +47.7±3.2 | -27.3±3.8 | +45.7±3.1 | -21.8±1.5 |
| 45 | 107.2±2.3 | 141.6±1.2 | 142.1±5.2 | 115.7±5.6 | 149.3±1.5 | 0.14±0.03 | 0.22±0.02 | 0.22±0.06 | 0.24±0.03 | 0.23±0.01 | -26.8±2.2 | +46.9±4.3 | -26.0±1.6 | +45.8±6.3 | -20.2±3.1 |
| 60 | 117.3±4.2 | 150.5±6.4 | 153.9±3.5 | 129.8±2.1 | 155.1±3.4 | 0.15±0.05 | 0.23±0.03 | 0.23±0.07 | 0.26±0.01 | 0.25±0.03 | -25.1±1.6 | +48.3±1.5 | -24.2±1.5 | +43.3±1.5 | -19.8±2.2 |
Comparative pharmacokinetic studies of prepared SMEs
All the prepared SMEs were compared for their pharmacokinetic parameters along with a suspension in male Wistar rats. The nimodipine, extracted from serum samples was estimated by a validated and reported HPLC method [25]. Serum concentration and time profiles of nimodipine upon oral administration are shown in Figure 3. The pharmacokinetic parameters are included in Table 7.
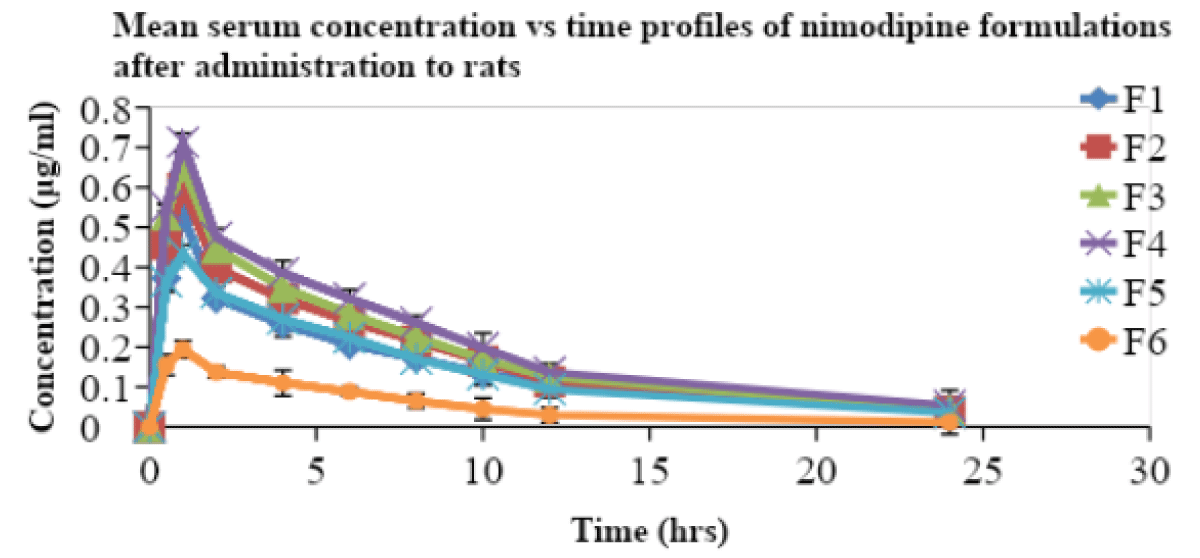
Figure 3: Serum concentration vs. time profiles of nimodipine from SMEs in rats.
| Table 7: Consolidated table showing the pharmacokinetic parameters of SMEs containing NM. | ||||||
| Parameter | F1 | F2 | F3 | F4 | F5 | F6 |
| Cmax (µg/ml) | 0.528 ± 0.03 | 0.598 ± 0.03 | 0.654 ± 0.041 | 0.713 ± 0.04* | 0.434 ± 0.02 | 0.194 ± 0.044 |
| tmax (h) | 1.0 ± 0 | 1.0 ± 0 | 1.0 ± 0 | 1.0 ± 0 | 1.0 ± 0 | 1.0 ± 0 |
| AUC total (µg/ml) h | 4.124 ± 0.24 | 4.647 ± 0.36 | 5.001 ± 0.33 | 5.666 ± 0.38* | 3.8131 ± 0.35 | 1.469 ± 0.260 |
| t½ (h) | 8.685 ± 0.51 | 7.178 ± 0.41 | 7.516 ± 1.13 | 7.974 ± 1.22 | 7.862 ± 1.20 | 8.448 ± 1.340 |
| MRT (h) | 11.303 ± 0.87 | 9.502 ± 1.31 | 9.580 ± 0.61 | 10.293 ± 1.07 | 10.108 ± 0.89 | 9.769 ± 1.304 |
| Note: *Significant at p < 0.001 when compared with F6 control. | ||||||
The Cmax and AUC total of F4 was very high when compared to that of F6 suspension. The Tmax of all the formulations was obtained at 1 hr. MRT and t½ for all formulations are not significantly different. The relative bioavailability of the SME formulations F1, F2, F3, F4, and F5 is found to be 2.8, 3.16, 3.4, 3.85, and 2.59 times that of suspension (F6) respectively. The oral bioavailabilities of nimodipine from the formulations (F1 to F5) were higher than those of suspension (F6). Negatively charged SME (F1) showed less oral bioavailability when compared to that of positively charged SME (F2). Similarly, F3 showed less oral bioavailability than F4. Positively charged SME (cationic) were reported [4] earlier to show better oral bioavailability due to increased uptake of globules by enterocytes. Similarly, F4, a positively charged SME containing tween 80 as co- emulsifier, a known P-gp inhibitor also increased the oral bioavailability. Here, no previous study reports are available for such a combination. This is due to the combination effect. This might also be due to the lymphatic transport of drugs [31,32] from SME formulations, resulting in a reduced first-pass effect. The design of F1, F2, F3, F4 formulations with inclusion of charge inducer and P-gp inhibitor produced higher oral bioavailability than that of suspension. P-gp, a drug efflux pump, is expressed in intestine and blood brain barrier [33,34]. Among (F1 -F4) formulations, F4 SME (positively charged with P-gp inhibitor) showed very much improved oral bioavailability than that of F1, F2, F3, and F5 formulations, and this might be due to the fact that positively charged SME had more electrostatic interactions with enterocytes in mucosal layer than negatively charged SMEs. Further, the tween 80, a known P-gp inhibitor might have also played a role in the improvement of oral bioavailability by reducing the efflux of NM drug. The statistical significance with paired t-test (p < 0.05) also showed that there was a significant difference in the bioavailability of F4 formulation when compared to that of F1, F2, F3, F5, and F6. This study showed that positively charged SME with a combination of P-gp inhibitor can be used for improved oral bioavailability of this lipophilic drug [4]. This strategy to enhance oral bioavailability is very simple to adopt in the pharmaceutical industry. And thus, the oral bioavailability of BCS class II drugs, like NM could be improved. The possibility of scale-up very much existed for this SME delivery system.
In this work, efforts were made to prepare stable nimodipine SMEs containing positive or negative charge inducer, with or without the combination of a P-gp inhibitor (tween 80) for oral bioavailability enhancement. As this drug NM was reported to exhibit low oral bioavailability (4% - 13%) due to its low solubility, extensive first-pass metabolism by CYP3A4 enzymes, and P-gp mediated efflux, here, SMEs were considered suitable and prepared by homogenization followed by the ultra-sonication method. All five SMEs were prepared with either a positive charge (stearyl amine) or negative charge (oleic acid) and also included or not include a P-gp inhibitor (Tween 80). A comparison was made with a plain control emulsion prepared without any added charge inducer and P-gp inhibitor. Characterization of SMEs was done by measuring size, polydispersity index, and zeta potential by zeta sizer. The prepared SMEs were found stable for two months at RT or 4 ○C. Further, the SMEs showed globules with a nano-size range. The rank order of oral bioavailability was F4 > F3 > F2 > F1 > F5 > F6. Nimodipine F4 SME formulation showed an increase in oral bioavailability by 3.85 fold due to overcoming the effects of first-pass metabolism by following lymphatic transport pathway, achieving P-gp inhibition and also due to positive charges on globules favoring more electrostatic interactions with enterocytes in mucosal surface for a better uptake than negatively charged SMEs. In the future, the developed NM SME (F4) could be produced in industry for the patients’ use.
The first author is thankful to the AICTE, New Delhi for providing financial assistance in the form of a Scholarship. We thank Dr Reddy’s laboratories, in Hyderabad, India for providing the gift sample of nimodipine.
- Porter CJ, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008 Mar 17;60(6):673-91. doi: 10.1016/j.addr.2007.10.014. Epub 2007 Nov 7. PMID: 18155801.
- Chakraborty S, Shukla D, Mishra B, Singh S. Lipid--an emerging platform for oral delivery of drugs with poor bioavailability. Eur J Pharm Biopharm. 2009 Sep;73(1):1-15. doi: 10.1016/j.ejpb.2009.06.001. Epub 2009 Jun 6. PMID: 19505572.
- Lundberg BB, Mortimer BC, Redgrave TG, Submicron lipid emulsion containing amphipathic poly ethylene glycol for use as drug carriers with prolonged circulation time. Int. J. Pharm. 1996; 134(1-2):119-127.
- Madishetty S, Syed MA, Prabhakar K, Kishan V. Cationic Diclofenac Lipid Nanoemulsion for Improved Oral Bioavailability. Int. J. Pharm.Sci and Nanotechnology. 2015; 8(2):2874- 2880.
- Prabhakar K, Afzal SM, Tween 80 containing lipid nanoemulsions for delivery of indinavir to brain, Acta Pharmaceutica Sinica B. 2013; 3(5):345- 353.
- Zhang H, Yao M, Morrison RA, Chong S. Commonly used surfactant, Tween 80, improves absorption of P-glycoprotein substrate, digoxin, in rats. Arch Pharm Res. 2003 Sep;26(9):768-72. doi: 10.1007/BF02976689. PMID: 14560928.
- Sun Y, Rui Y, Wenliang Z, Tang X. Nimodipine semi-solid capsules containing solid dispersion for improving dissolution. Int J Pharm. 2008 Jul 9;359(1-2):144-9. doi: 10.1016/j.ijpharm.2008.03.040. Epub 2008 Apr 7. PMID: 18499371.
- Kale AA, Patravale VB. Design and evaluation of self-emulsifying drug delivery systems (SEDDS) of nimodipine. AAPS PharmSciTech. 2008;9(1):191-6. doi: 10.1208/s12249-008-9037-9. Epub 2008 Feb 5. PMID: 18446481; PMCID: PMC2976914.
- Chalikwar SS, Belgamwar VS, Talele VR, Surana SJ, Patil MU. Formulation and evaluation of Nimodipine-loaded solid lipid nanoparticles delivered via lymphatic transport system. Colloids Surf B Biointerfaces. 2012 Sep 1;97:109-16. doi: 10.1016/j.colsurfb.2012.04.027. Epub 2012 Apr 25. PMID: 22609590.
- Sun C, Wang J, Liu J, Qiu L, Zhang W, Zhang L. Liquid proliposomes of nimodipine drug delivery system: preparation, characterization, and pharmacokinetics. AAPS PharmSciTech. 2013 Mar;14(1):332-8. doi: 10.1208/s12249-013-9924-6. Epub 2013 Jan 15. PMID: 23319300; PMCID: PMC3581684.
- Nguyen TT, Duong VA, Maeng HJ. Pharmaceutical Formulations with P-Glycoprotein Inhibitory Effect as Promising Approaches for Enhancing Oral Drug Absorption and Bioavailability. Pharmaceutics. 2021 Jul 20;13(7):1103. doi: 10.3390/pharmaceutics13071103. PMID: 34371794; PMCID: PMC8309061.
- Zhang Q, Jiang X, Jiang W, Lu W, Su L, Shi Z. Preparation of nimodipine-loaded microemulsion for intranasal delivery and evaluation on the targeting efficiency to the brain. Int J Pharm. 2004 May 4;275(1-2):85-96. doi: 10.1016/j.ijpharm.2004.01.039. PMID: 15081140.
- Yu J, He HB, Tang X. Formulation and evaluation of nimodipine-loaded lipid microspheres. J Pharm Pharmacol. 2006 Nov;58(11):1429-35. doi: 10.1211/jpp.58.11.0002. PMID: 17132204.
- Barmpalexis P, Kanaze FI, Georgarakis E. Developing and optimizing a validated isocratic reversed-phase high-performance liquid chromatography separation of nimodipine and impurities in tablets using experimental design methodology. J Pharm Biomed Anal. 2009 Jul 12;49(5):1192-202. doi: 10.1016/j.jpba.2009.03.003. Epub 2009 Mar 20. PMID: 19369025.
- Krishna G, Wood GC, Sheth BB. Improving emulsification efficacy of lecithin by formulation design. I: Effect of adding a secondary surfactant. PDA J Pharm Sci Technol. 1998 Nov-Dec;52(6):331-6. PMID: 10050132.
- Baranda AB, Etxebarria N, Jiménez RM, Alonso RM. Development of a liquid-liquid extraction procedure for five 1,4-dihydropyridines calcium channel antagonists from human plasma using experimental design. Talanta. 2005 Oct 31;67(5):933-41. doi: 10.1016/j.talanta.2005.04.028. Epub 2005 May 31. PMID: 18970261.
- Andrew JH, William NC. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv. Drug Deliv. Rev. 1997; 25(1):103-128.
- O'Driscoll CM. Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci. 2002 Jun;15(5):405-15. doi: 10.1016/s0928-0987(02)00051-9. PMID: 12036717.
- Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008 Mar 17;60(6):625-37. doi: 10.1016/j.addr.2007.10.010. Epub 2007 Nov 4. PMID: 18068260.
- Washington C. Stability of lipid emulsion for drug delivery. Adv. Drug Deliv. Rev. 1996; 20(2-3):131-145.
- Chidambaram N, Burgess DJ. A novel in vitro release method for submicron sized dispersed systems. AAPS PharmSci. 1999;1(3):E11. doi: 10.1208/ps010311. PMID: 11741207; PMCID: PMC2761125.
- Gribar JJ, Ramachandra M, Hrycyna CA, Dey S, Ambudkar SV. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J Membr Biol. 2000 Feb 1;173(3):203-14. doi: 10.1007/s002320001020. PMID: 10667916.
- Chen H, Khemtong C, Yang X, Chang X, Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discov Today. 2011 Apr;16(7-8):354-60. doi: 10.1016/j.drudis.2010.02.009. Epub 2010 Mar 3. PMID: 20206289.
- Ishii F, Sasaki I, Ogata H. Effect of phospholipid emulsifiers on physicochemical properties of intravenous fat emulsions and/or drug carrier emulsions. J Pharm Pharmacol. 1990 Jul;42(7):513-5. doi: 10.1111/j.2042-7158.1990.tb06609.x. PMID: 1980297.
- Arimoto I, Handa T. Lipid emulsion: Formation, stability and metabolism, Encyclopedia of Surface and Colloid Sci. 1998;13(3):3087-3097.
- Yáñez JA, Wang SW, Knemeyer IW, Wirth MA, Alton KB. Intestinal lymphatic transport for drug delivery. Adv Drug Deliv Rev. 2011 Sep 10;63(10-11):923-42. doi: 10.1016/j.addr.2011.05.019. Epub 2011 Jun 13. PMID: 21689702; PMCID: PMC7126116.
- Gutierrez JM, Gonzalez C, Maestro A, Sole I. Nano-emulsions : New applications and optimization of their preparation. Current opinion in Colloid and Interface Sci. 2008; 13:245-351.
- Demeule M, Régina A, Jodoin J, Laplante A, Dagenais C, Berthelet F, Moghrabi A, Béliveau R. Drug transport to the brain: key roles for the efflux pump P-glycoprotein in the blood-brain barrier. Vascul Pharmacol. 2002 Jun;38(6):339-48. doi: 10.1016/s1537-1891(02)00201-x. PMID: 12529928.
- Trevaskis NL, Charman WN, Porter CJ. Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev. 2008 Mar 17;60(6):702-16. doi: 10.1016/j.addr.2007.09.007. Epub 2007 Nov 7. PMID: 18155316; PMCID: PMC7103284.
- Pathak R, Dash RP, Misra M, Nivsarkar M. Role of mucoadhesive polymers in enhancing delivery of nimodipine microemulsion to brain via intranasal route. Acta Pharm Sin B. 2014 Apr;4(2):151-60. doi: 10.1016/j.apsb.2014.02.002. Epub 2014 Apr 2. PMID: 26579378; PMCID: PMC4590727.
- Takamura A, Ishii F, Noro S, Tanifuji M, Nakajima S. Study of intravenous hyperalimentation: effect of selected amino acids on the stability of intravenous fat emulsions. J Pharm Sci. 1984 Jan;73(1):91-4. doi: 10.1002/jps.2600730124. PMID: 6694093.
- Varshika E, Prabhakar K, Kishan V. Preparation, characterization, and in vivo pharmacodynamic evaluation of parenteral diclofenac submicron lipid emulsions. PDA J Pharm Sci Technol. 2009 Sep-Oct;63(5):380-9. PMID: 20158044.
- Teng Z, Yu M, Ding Y, Zhang H, Shen Y, Jiang M, Liu P, Opoku-Damoah Y, Webster TJ, Zhou J. Preparation and characterization of nimodipine-loaded nanostructured lipid systems for enhanced solubility and bioavailability. Int J Nanomedicine. 2018 Dec 21;14:119-133. doi: 10.2147/IJN.S186899. PMID: 30613141; PMCID: PMC6306054.
- He Z, Zhong D, Chen X, Liu X, Tang X, Zhao L. Development of a dissolution medium for nimodipine tablets based on bioavailability evaluation. Eur J Pharm Sci. 2004 Mar;21(4):487-91. doi: 10.1016/j.ejps.2003.11.009. PMID: 14998579.