
More Information
Submitted: March 29, 2023 | Approved: April 06, 2023 | Published: April 07, 2023
How to cite this article: Cui J, Qiaerxie G, Qin H, Long F, Wang X, et al. Design and optimization of mRNAs encoding an Anti-TIGIT antibody with therapeutic potential for cancer in TIGIT-humanized BALB/c Mice. Arch Pharm Pharma Sci. 2023; 7: 008-016.
DOI: 10.29328/journal.apps.1001038
Copyright License: © 2023 Cui J, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: TIGIT; Antibody; mRNA; Optimization; Lipid nanoparticle; Cancer immunotherapy

Design and optimization of mRNAs encoding an Anti-TIGIT antibody with therapeutic potential for cancer in TIGIT-humanized BALB/c Mice
Jingmin Cui1,2#, Gulisaina Qiaerxie2,4#, Hui Qin2,3, Feng Long1,4, Xi Wang2, Zhixin Yang2, Peng Du2* and Yong Cui1*
1School of Medical Device, Shenyang Pharmaceutical University, Liaoning, China
2Beijing Institute of Biotechnology, Beijing, China
3School of Pharmacy, North Sichuan Medical College, Sichuan, China
4Department of Pharmacy, Maternal and Child Health Care Hospital of Zaozhuang, Shandong, China
#These authors contributed equally to this work
*Address for Correspondence: Peng Du, Beijing Institute of Biotechnology, Beijing, China, Email: [email protected]
mRNA drugs are synthesized using cell-free systems without complex and stringent manufacturing processes, which makes their preparation simple, efficient, and economical. Over the past few years, mRNAs encoding antibodies have been one of the research frontiers of antibody drug development. In cancer immunotherapy, mRNAs encoding immune checkpoint antibodies may be advantageous regarding antibody persistence and durability of the anti-tumor immune response of patients. In our previous study, a candidate antibody—AET2010—targeting the novel immune checkpoint TIGIT was reported. Its anti-tumor activity was also investigated using adoptive transfer of NK-92MI cells in a xenograft mouse model, but the limitations of the model did not facilitate precise evaluation. In the present study, we further investigated the therapeutic potential of AET2010 for cancer in TIGIT-humanized BALB/c mice. Next, we explored the design, synthesis, and optimization of mRNAs encoding AET2010 and ultimately obtained a candidate mRNA (mRNA-BU) with favorable in vitro and in vivo expression levels of active AET2010. Particularly, lipid-nanoparticle-encapsulated mRNA-BU delivered to mice produced AET2010 with significantly higher peak concentration and expression duration than an equivalent dose of original AET2010. This study provides a sound basis for developing novel drugs targeting TIGIT.
Mature mRNAs consist of five structural elements; from 5′ to 3′, these are the cap structure, 5′-untranslated region (UTR), open reading frames (ORFs), 3′ UTR, and poly-A tail (a chain of adenine nucleotides), respectively. The cap structure and poly-A tail protect the mRNA from exonuclease degradation and improve its stability and translation efficiency [1,2]. The UTRs are responsible for regulating translation and protein expression and affect the translation efficiency, half-life, maximum expression level, and stability of the mRNA [3,4]. The translatability of the ORFs is also crucial to the mRNA [5-7]. The in vivo use of in vitro-transcribed (IVT) mRNAs for protein expression dates back to the 1990s [8,9]. A series of design and modification technologies for mRNA synthesis has improved the biological characteristics of IVT mRNAs [1,10,11], thus improving the stability of mRNA molecules and the translation level of proteins. However, mRNAs are negatively charged molecules with a relatively large molecular weight (104–106 Da), which makes it difficult to traverse the anionic lipid bilayer of cells and are easily degraded. Thus, there exist two barriers to mRNA delivery into the cell: enzymatic degradation during delivery and transport across the membrane barrier due to electrostatic repulsion [12]. During the past few years, many innovative solutions based on materials have been developed; the most widely used appears to be the nano-delivery systems. Lipid nanoparticles (LNPs) are biocompatible carriers with phospholipid monolayers that encapsulate mRNA, preventing degradation and allowing efficient delivery [13,14]. The gradual resolution of these technical problems makes it possible to synthesize mRNAs in vitro as a therapeutic drug. Specifically, during the coronavirus disease 2019 pandemic, mRNA drug development has been accelerated. The novel coronavirus mRNA vaccine mRNA-1273 developed by Moderna and BNT162b2 jointly developed by Pfizer and BioNTech have been authorized for emergency use, which showed a promising prospect for mRNA drugs [15].
Therapeutic antibodies constitute the primary type of biological drug. However, the complex and stringent manufacturing processes of antibody drug development have hindered patient access to a certain extent. Using mRNA-encoded antibodies may overcome some challenges in the production of antibody drugs. mRNA drugs are biological macromolecules synthesized by a cell-free system and thus do not depend on complex technological processes such as cell culture, scale fermentation, and protein purification, making the production of antibody-encoding mRNAs simple, efficient, and economical [1,16,17]. In addition, in vivo administration of antibody-encoding mRNAs would facilitate the delivery of therapeutic antibodies to the body continuously for a long period, reducing the required frequency of dosing. To date, no drugs consisting of IVT mRNAs encoding antibodies are available in the market; however, significant advances have been made—for example, a successful phase I clinical trial (NCTO3829384) of mRNA encoding a neutralizing antibody to Chikungunya virus (CHIKV) has been completed by Moderna [17]. In cancer immunotherapy, treatment with mRNAs encoding immune checkpoint antibodies may be advantageous in terms of antibody persistence and durable anti-tumor immune response of patients.
TIGIT is a novel immune checkpoint molecule expressed in T cells, natural killer (NK) cells, and other immune cells. Its immunomodulatory effects on the function of immune cells make it an attractive candidate drug target. By blocking the signal pathway between TIGIT and its receptors, the exhaustion of tumor-infiltrating NK cells and CD8+ T cells may be reversed, and the anti-tumor immunity of patients may be restored. [18-20]. In a previous study, we obtained a candidate therapeutic antibody—AET2010—targeting TIGIT. The anti-tumor activity of AET2010 was investigated in a xenograft mouse model with an adoptive transfer of NK-92MI cells. However, this model was not sufficient for the precise evaluation of the anti-tumor efficacy of AET2010 because the effect of NK cells was boosted, and the potential anti-tumor effect of CD8+ T cells was overlooked [21].
In the present study, we further investigated and ascertained the therapeutic potential of AET2010 for cancer in TIGIT-humanized BALB/c mice. Subsequently, we explored the design, synthesis, and optimization of mRNAs, and their expression in mice by LNP delivery to obtain a candidate mRNA that could effectively encode AET2010. Totally, our study aimed to provide a sound basis for the development of novel drugs targeting TIGIT.
Antibody preparation
The genes encoding the variable regions of light and heavy chains of RG6058 (tiraglumab) from patent US10047158B2 were synthesized and cloned into expression vectors pABκ and pABG (maintained in our laboratory) respectively. HEK-293F cells were cultured at 37 °C in FreeStyleTM 293 expression medium (Gibco, NY, USA) and incubated at 37 °C in a 5% CO2 incubator. For antibody expression, Human embryonic kidney 293 cells (HEK-293F) were transfected with equal quantities of heavy chain and light chain expression plasmids using FectoPRO transfection reagent (Polyplus‐Transfection SA, FR) and cultured at 37 °C and 5% CO2 while being shaken at 125 rpm for 4 days; the cell cultures were centrifuged at 8000 rpm for 10 min to collect the culture supernatant, which was filtered through a 0.45 µm membrane. Antibodies secreted in the supernatant were purified using HiTrap™ Mabselect SuRe™ (Cytiva, 11003493, USA), replacing 0.1 M citric acid buffer (pH 3.0) with phosphate-buffered saline (PBS; pH 7.4) using HiTrapTM desalting columns (Cytiva, 29048684, USA). A spectrophotometer (Thermo Nanodrop One C, NY, USA) was then used to quantify protein concentration, and the presence of the protein was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis.
Surface plasmon resonance assay
The interaction affinity between antibodies and recombinant human TIGIT-His (SinoBiological, Beijing, China) was calculated on the basis of kinetic constants using the BiacoreTM 3000 system. Multi-cycle kinetics analysis was used for kinetic analysis experiments. Purified AET2010, RG6058, and MK7684 (vibostolimab) were captured on a CM5 chip using the Human Antibody Capture Kit (Cytiva, USA). Recombinant human TIGIT-His with a concentration ranging from 2.05 nM to 80.00 nM in HBS-EP+ running buffer was passed over the chip at a rate of 30 µL/min at 25 °C. A 3 min association period was followed by a 6 or 12 min dissociation period. A cycle of repeated measurements was set for quality control, and two cycles of blank controls were used to correct system deviations. The regeneration step consisted of chip flushing with 3 M MgCl2l for 30 s at the rate of 30 µL/min. The experimental data were fit to a 1:1 binding model using BIAcore™ 3000 evaluation software. Equilibrium dissociation constants (KD) were calculated according to fitting curves.
Mice and animal experiments
Female TIGIT-humanized BALB/c mice (BALB/c-hTIGIT, 6–7 weeks old) were purchased from Gempharmatech (T004023, Jiangsu, China). All mice were maintained under specified pathogen-free (SPF) conditions and the animal use protocol was approved by the Animal Ethics Committee of Gempharmatech Co. Ltd. (GPTAP20211203-1). To establish a xenograft tumor model, the mice were subcutaneously inoculated with 100 µL of CT26 colorectal cancer cells suspended in PBS (5 × 105 cells per mouse). Tumor dimensions were measured twice weekly, and tumor volume was calculated as follows: volume = 0.5 × long diameter × (short diameter)2. The mice were randomized into four groups when the tumor volume reached 80–100 mm3. The day of grouping was defined as D0. On D0, D3, D7, D10, D14 and D17, mice were intraperitoneally injected with AET2010, RG6058, MK7684, and isotype human IgG (10 µg/g of mouse body weight). Mouse body weight and tumor volume were measured at D0, D2, D5, D9, D12, D16 and D19.
Relative tumor volume on day t for mouse number n (RTVn) was calculated as follows:
RTVn = Vnt/Vn0
Where Vnt indicates tumor volume on day t for mouse number n, and Vn0 indicates tumor volume on D0 for mouse number n.
The tumor growth inhibition (TGI) rate was calculated as follows:
TGI = (1 - (mean RTVtreat)/(mean RTVcontrol)) × 100%
Where mean RTVtreat indicates the mean RTV of a treatment group, and mean RTVcontrol indicates the average RTV of the isotype human IgG group.
mRNA preparation
Gene sequences containing the T7 promoter, 5′ UTR, ORF (antibody-coding sequence), and 3′ UTR sequences were synthesized and cloned into pMV plasmid vectors to construct DNA templates. The mRNAs were synthesized in vitro using T7 RNA polymerase-mediated (Vazyme, Nanjing, China) transcription of DNA templates linearized by digestion with XbaI (NEB, USA). Transcribed samples were purified using magnetic beads (Vazyme, Nanjing, China). A 7-methyl guanosine cap (m7Gppp, Cap 0) was added to the 5′ ends of RNA using a vaccinia virus capping system (Vazyme, Nanjing, China), and then the cap was 2′-O-methylated to form Cap 1. The poly-A tail was added to the 3′UTR using Escherichia coli Poly (A) polymerase (NEB, USA). The prepared mRNAs were verified by agarose gel electrophoresis.
Cell culture and antibody expression
Chinese Hamster Ovary cells (CHO) and African green monkey SV40 transformed kidney cells (COS-7) (maintained in our laboratory) were incubated at 37 °C in Dulbecco’s modified Eagle’s minimal essential medium (DMEM) (Procell, Wuhan, China) containing 10% fetal bovine serum (Gibco), 100 U/mL of penicillin, and 100 mg/mL of streptomycin (Gibco). Human Embryonic Kidney 293 Cells (HEK-293F) were incubated at 37 °C in FreeStyleTM 293 expression medium (Gibco).
CHO, COS-7 and HEK-293F cells were seeded in 48-well plates at 100,000 cells/well. After 24 h, cells were transfected with 0.9 µg of mRNA using 2.7 µL of Lipofectamine™ MessengerMAX™ transfection reagent (Thermo Fisher Scientific, MA, USA). The mass ratio of light chain to heavy chain in duplex mRNA was 2:1. The supernatant was collected by centrifuging at 8000 rpm for 10 min at 48 h post-transfection and analyzed by enzyme-linked immunosorbent assay (ELISA). The cell cultures were centrifuged at 8000 rpm for 10 min to collect the culture supernatant
Enzyme-linked immunosorbent assay
mRNA-encoded antibody proteins expressed in vitro and in vivo were quantified using ELISA. The concentration of antibody proteins was calculated on the basis of a standard curve. For ELISA, 96-well microplates (Corning, NY, USA) were coated with recombinant human TIGIT-His (prepared in our laboratory; 100 ng per well) and incubated overnight at 4 °C and then blocked with PBS containing 2.5% fat-free powdered milk for 1 h at 37 °C. A standard curve was plotted from serial dilutions (1.465–1000.000 ng/mL) of the original AET2010 in PBS buffer containing 2.5% fat-free powdered milk and 0.1% Tween 20. Cell expression supernatant or mouse serum was serially diluted and then incubated for 1 h at 37 °C. After washing, horseradish peroxidase-conjugated goat anti-human IgG (ZSGB-Bio, Beijing, China) was added to the mixture and incubated for 45 min at 37 °C. The absorbance of the resultant mixture at 450 nm and 595 nm in 3, 3’, 5, 5’-Tetramethylbenzidine (TMB) substrate was measured (Thermo Multiskan MK3).
Codon optimization
The distribution of full-length sequence codon usage bias was analyzed using the MinMax% tool [22]. Global codon substitution was performed using the human codon usage bias table (GenBank_63, http://www.kazTsa.or.jp/codon/) and minimum free energy (MFE) of sequences was analyzed using RNA structure (http://rna.urmc.rochester.edu/RNAstructureWeb/) [23,24]. The guanine and cytosine content (GC%) and codon adaptation index (CAI) values were estimated. The secondary structure of mRNA was predicted using LinearDesign Web Server (beta) (http://rna.baidu.com/).
Lipid nanoparticle formulation
SM-102, DSPC, cholesterol, and mPEG-DMG-2K at the molar ratio of 50:10:38.5:1.5 were dispersed in absolute ethanol, shaken well, and stored at 4 °C. Next, 1 mg of mRNA was added to sodium acetate buffer, and then the organic phase mixture and mRNA–sodium acetate solution were added to the two inlets of the INano™ L nanomedicine preparation system in a volumetric ratio of 3:1. Liposomal nanoparticles were prepared at a flow rate of 12 mL/min. After ultrafiltration and solution exchange, the LNP-encapsulated mRNA was dispersed in 10× PBS. Particle size was characterized using a Zetasizer Nano ZS to obtain the average particle size and the polydispersity index (PI). The product was then diluted or concentrated to obtain the required concentrations, and aliquots of the solution were stored at 4 °C.
In vivo mRNA expression
Female BALB/c mice (6–8 weeks old) (Beijing Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) were randomly divided into six groups (n = 3 per group). Mice were intravenously administered with 150 µg of the original AET2010 or one of the mRNA-LNPs; blank LNPs served as the negative control group. Mouse serum samples were collected at 2, 6, 12, 24, 48, 72, and 96 h after administration of treatment and stored at −80 °C for subsequent testing. Serum antibody concentration was assessed using ELISA, as described above. The animal use protocol was approved by the Animal Ethics Committee of the Academy of Military Medical Sciences (No. IACUC-DWZX-2021-032).
Statistical analysis
In animal experiments, all data were presented as mean ± standard deviation (SD), and statistical analyses were performed using GraphPad Prism software (version 8.0). The differences between the groups were analyzed by a two-tailed Student’s t-test and considered statistically significant if p < 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001). The quantitated concentrations of AET2010 were presented as mean ± SD and the data were analyzed using GraphPad Prism software.
AET2010 showed therapeutic potential for cancer in TIGIT-humanized BALB/c mice
We synthesized AET2010 and two control antibodies RG6058 and MK7684 and studied the binding activity of the three antibodies to TIGIT using a surface plasmon resonance assay. Compared with the two control antibodies, AET2010 showed a weaker affinity of 2.92 nM (Figure 1A-1C), which was consistent with our previous findings [21]. Next, we established a xenograft tumor model in TIGIT-humanized BALB/c mice with subcutaneous inoculation of CT26 colorectal cancer cells and assessed the anti-tumor efficacy of the antibodies. The results (Figure 1D) showed that AET2010 displayed significant tumor proliferation inhibitory activity with a TGI rate of 50.19%, which was slightly lower than that of RG6058 (69.41%) and MK7684 (62.67%), but there was no significant difference observed between anti-TIGIT antibody groups. Thus, the therapeutic potential of AET2010 for cancer was further confirmed using a precise and favorable model.
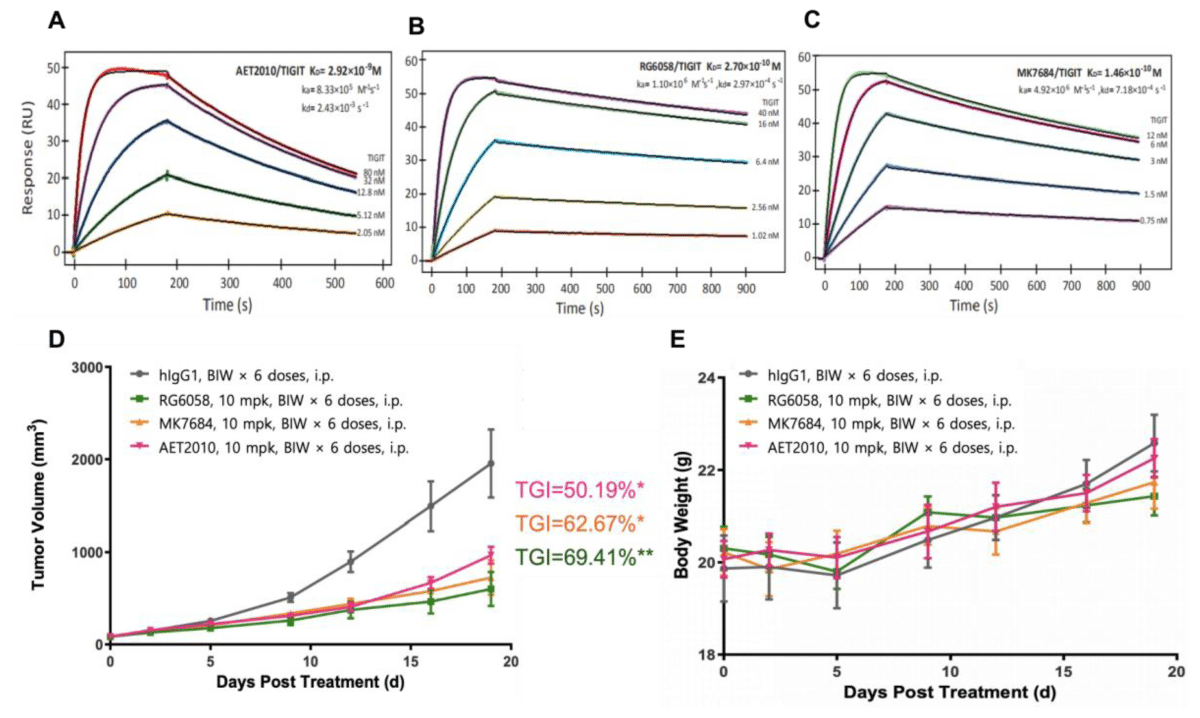
Figure 1: Representative sensorgrams of AET2010 (A), RG6058 (B), and MK7684 (C) interacting with human TIGIT in surface plasmon resonance assays and their anti-tumor efficacy in TIGIT-humanized BALB/c-hTIGIT mice (D, E). The antibodies were immobilized on the sensor chip surface, and human TIGIT-His at serially diluted concentrations was passed over the chip. The fitting curves (thin black lines) were derived from the analysis using the 1:1 binding mode. Equilibrium disassociation constants (KD) were calculated according to the fitting curves. KD = kd/ka; ka, association rate constant; kd, disassociation rate constant. (A) AET2010, KD = 2.92 nM; (B) RG6058, KD = 0.27 nM; (C) MK7684, KD = 0.146 nM. Changes in tumor volume (D) and body weight (E) signifying the anti-tumor efficacy of antibodies in BALB/c-hTIGIT mice subcutaneously inoculated with CT26 cells. TGI, tumor growth inhibition; mpk, milligram per kilogram; BIW, twice a week; i.p., intraperitoneal injection. Data were analyzed using GraphPad Prism Software, and data are presented as mean ± standard deviation. ***p < 0.001, **p < 0.01, *p < 0.05 (unpaired t-test); n = 6.
Design, synthesis and in vitro expression of mRNAs encoding AET2010
Because canonical antibodies comprise two heavy chains and two light chains, we designed two different types of mRNA encoding AET2010: one mRNA with a single ORF encoding the heavy and light chain of AET2010 (mRNA-S) (Figure 2A) and two mRNAs and ORFs encoding the heavy and light chains separately (mRNA-B) (Figure 2B). For mRNA-S, the heavy chain and light chain were separated by the gene of 2A “self-cleaving” peptides (P2A), which promotes P2A-mediated ribosomal skipping followed by host furin-mediated cleavage. The codons encoding amino acids of AET2010 were consistent with those of the original antibody expressed in the FreeStyle™ 293 system. Cap 1 was selected as the cap structure for both types of mRNAs. The 5′ UTR was derived from human α-globin RNA with an optimized Kozak consensus sequence. Sequences containing two sequence elements derived from the partial amino-terminal enhancer of split (AES) mRNA and the mitochondrial-encoded 12S rRNA were selected as the 3′ UTR [25]. The plasmids were synthesized and constructed as per the design, followed by in vitro transcription, capping, and tailing modification. Ultimately, we obtained the two types of mRNA samples (Figure 3A).
To confirm the mRNA expression of AET2010 in different cells, mRNAs were transfected into CHO, COS-7, and HEK-293F cells cultured in vitro. The concentration of AET2010 protein in the expression supernatant was quantified by ELISA. mRNA-B exhibited a higher expression level of AET2010 than mRNA-S in the three cell lines (Figure 3C).
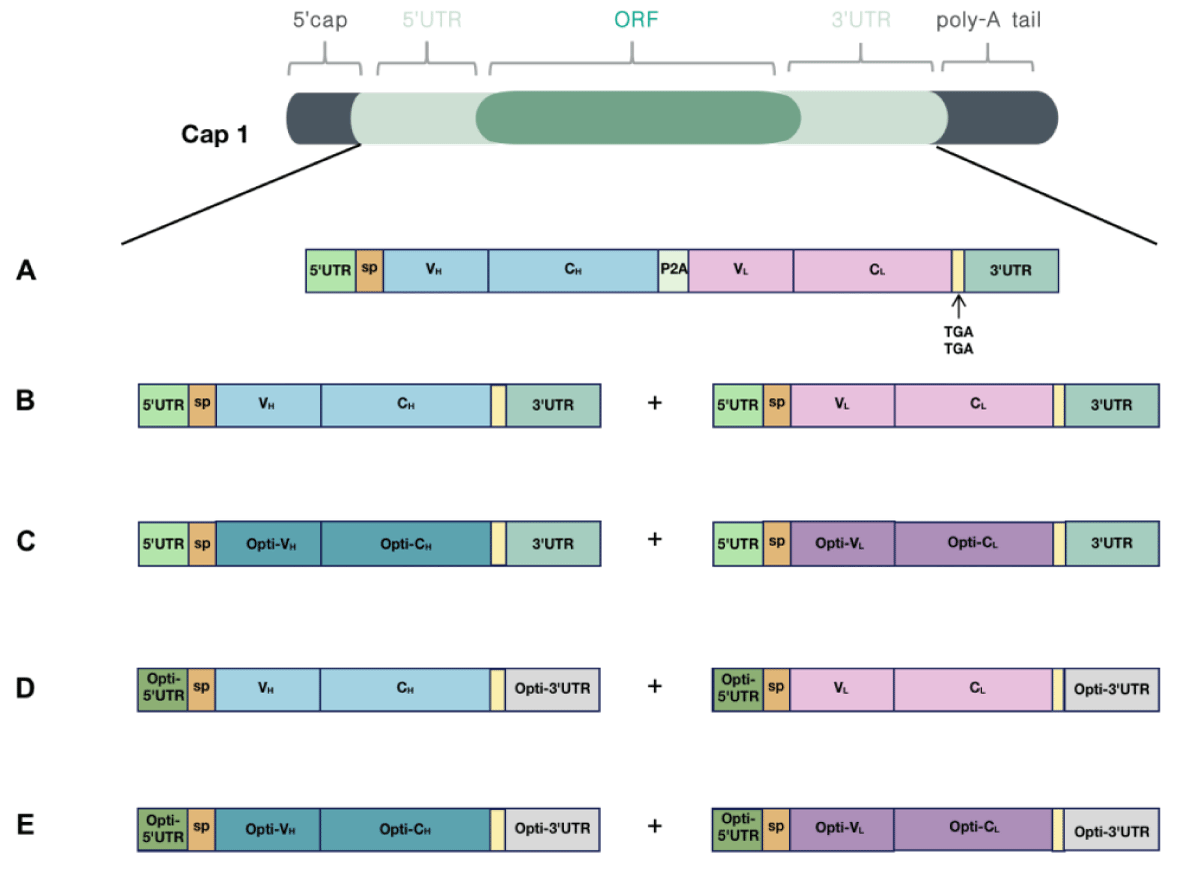
Figure 2: Design and optimization of mRNAs encoding AET2010. Cap 1 was selected as the cap structure for both types of mRNAs; the poly-A tail was added to the mRNA tail by polymerase after the DNA was transcribed into mRNA. (A) The encoding region of light and heavy chains was contained in a single ORF (mRNA-S). The 5′ UTR was derived from human α-globin RNA with an optimized Kozak sequence, and the 3′ UTR contained two sequence elements derived from the partial amino-terminal enhancer of split (AES) mRNA and the mitochondrial-encoded 12S ribosomal RNA. The encoding region of light and heavy chains was separated by the 2A “self-cleaving” peptides (P2A) gene. (B) The encoding region of light and heavy chains were contained in two separate ORFs (mRNA-B), and the 5′ UTR and 3′ UTR sequences matched those of mRNA-S. (C-E) Optimized mRNAs based on mRNA-B. (C) The codons of the light chain and heavy chain were optimized (mRNA-BC). (D) The 5′ UTR was substituted with UTR2 with high ribosome loading derived from the NCBI Gene Expression Omnibus database (the original library ID of UTR2: 317915), and the 3′ UTR was replaced by two copies of human β-globin mRNA (HBB gene) in tandem (mRNA-BU). (E) Both codon optimization and UTR substitution were performed (mRNA-BCU).
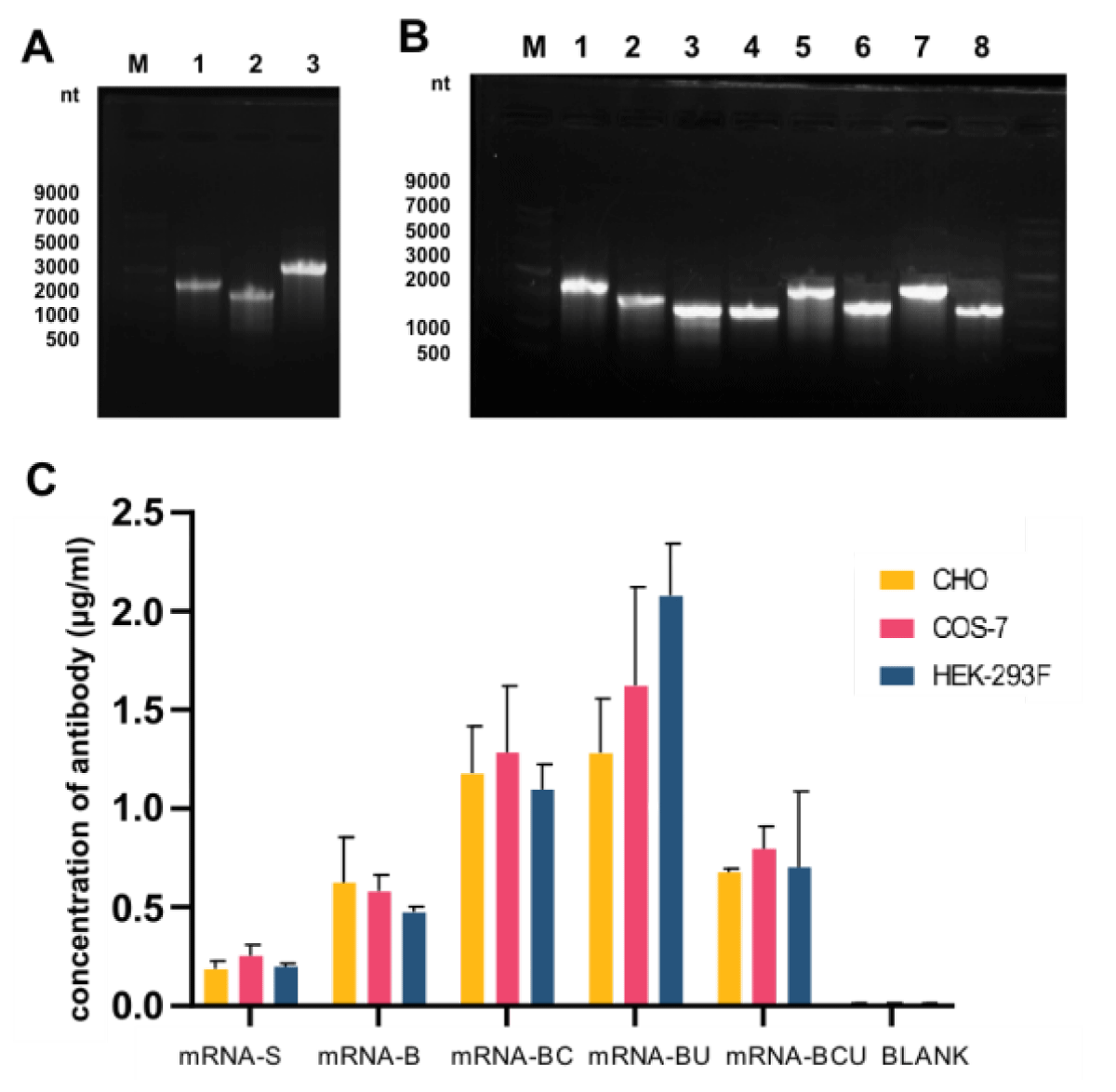
Figure 3: Preparation and in vitro expression of mRNAs. (A) Agarose gel electrophoresis of prepared mRNA-S (Lane 3), the heavy chain of mRNA-B (Lane 1), and the light chain of mRNA-B (Lane 2). Lane M: single-stranded RNA (ssRNA) ladder. (B) Agarose gel electrophoresis of optimized mRNAs. Lane M: ssRNA ladder, 1: the heavy chain of mRNA-B, 2: the light chain of mRNA-B, 3: the heavy chain of mRNA-BC, 4: the light chain of mRNA-BC, 5: the heavy chain of mRNA-BU, 6: the light chain of mRNA-BU, 7: the heavy chain of mRNA-BCU, and 8: the light chain of mRNA-BCU. (C) In vitro expression levels of AET2010 by synthesized mRNAs transfected into CHO, COS-7, and HEK-293 cells. CHO, Chinese Hamster Ovary cell; COS-7, African green monkey SV40 transformed kidney cell; HEK-293, Human embryonic kidney 293 cells.
Optimization and verification of mRNAs encoding AET2010
To improve the expression efficiency, we attempted to further optimize mRNA-B. First, the codon optimization of the ORF region was performed. The preferred human codon bias was used, resulting in a higher MinMax% distribution exceeding 60% (Figure 4). The ratio of guanine and cytosine (GC%) and codon adaptation index (CAI) values were also improved. However, the MFE of the sequence slightly increased, which may decrease the stability of the mRNA structure (Table 1). Subsequently, we performed pairwise substitution of the 5′ UTR and 3′ UTR, which affected the stability and translation efficiency of mRNA. A different 5′ UTR sequence with high ribosome loading derived from the NCBI Gene Expression Omnibus database (UTR2; the original library ID of UTR2 is 317915) [26] and 3′ UTR containing two copies of human β-globin mRNA (HBB gene) in tandem [26,27] were applied. The mRNA-B with codon optimization, UTR substitution, or both were named mRNA-BC (Figure 2C),
| Table 1: MFE, GC% and CAI values of the open reading frame sequence of mRNA-B before and after codon optimization. | |||
| Sequence | MFE | GC% | CAI |
| Before optimization | -835.4 | 0.582 | 0.773 |
| After optimization | -744.4 | 0.612 | 0.938 |
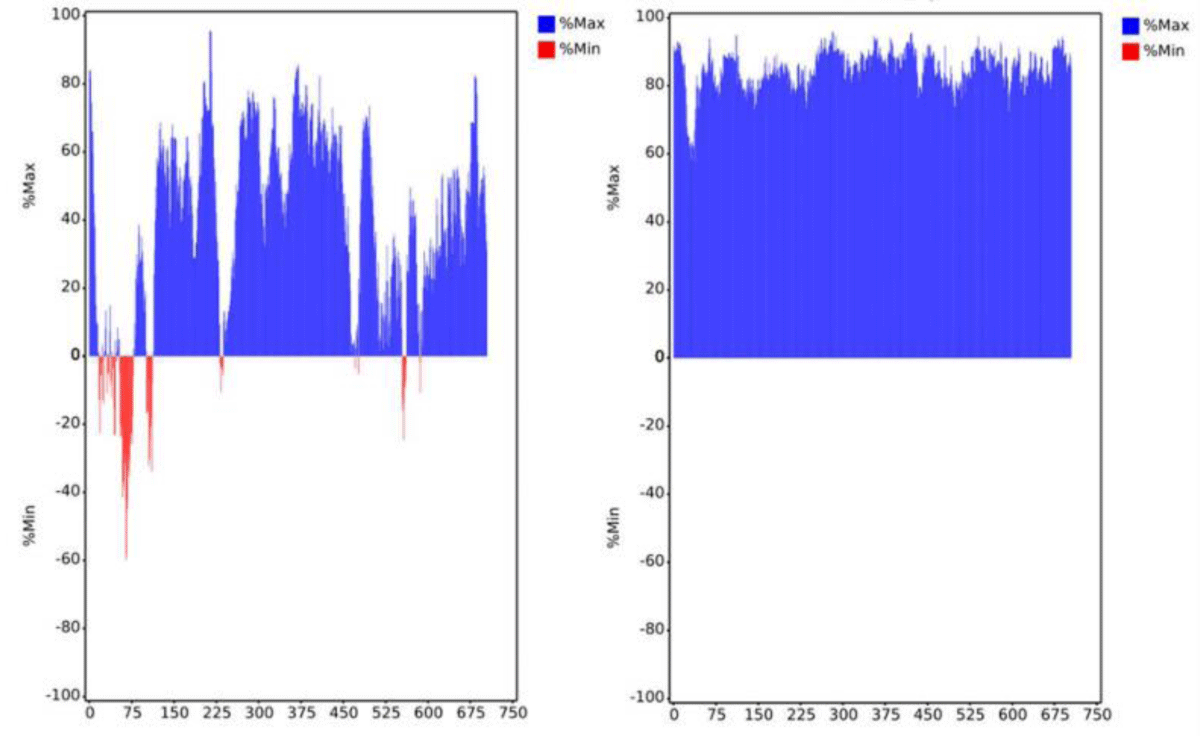
Figure 4: Distribution of codon usage bias of mRNA-B open reading frame sequence by MinMax% before (left panel) and after (right panel) codon optimization. The horizontal axis shows the amino acid sequence number of the heavy chain and light chain, respectively. Min% represents the proportion of rare codons; Max% represents the proportion of favorable codons.
mRNA-BU (Figure 2D) and mRNA-BCU (Figure 2E), respectively. The secondary structure and stability of optimized mRNAs were preliminarily predicted, indicating that the stability changes were not significant (Figure S1).
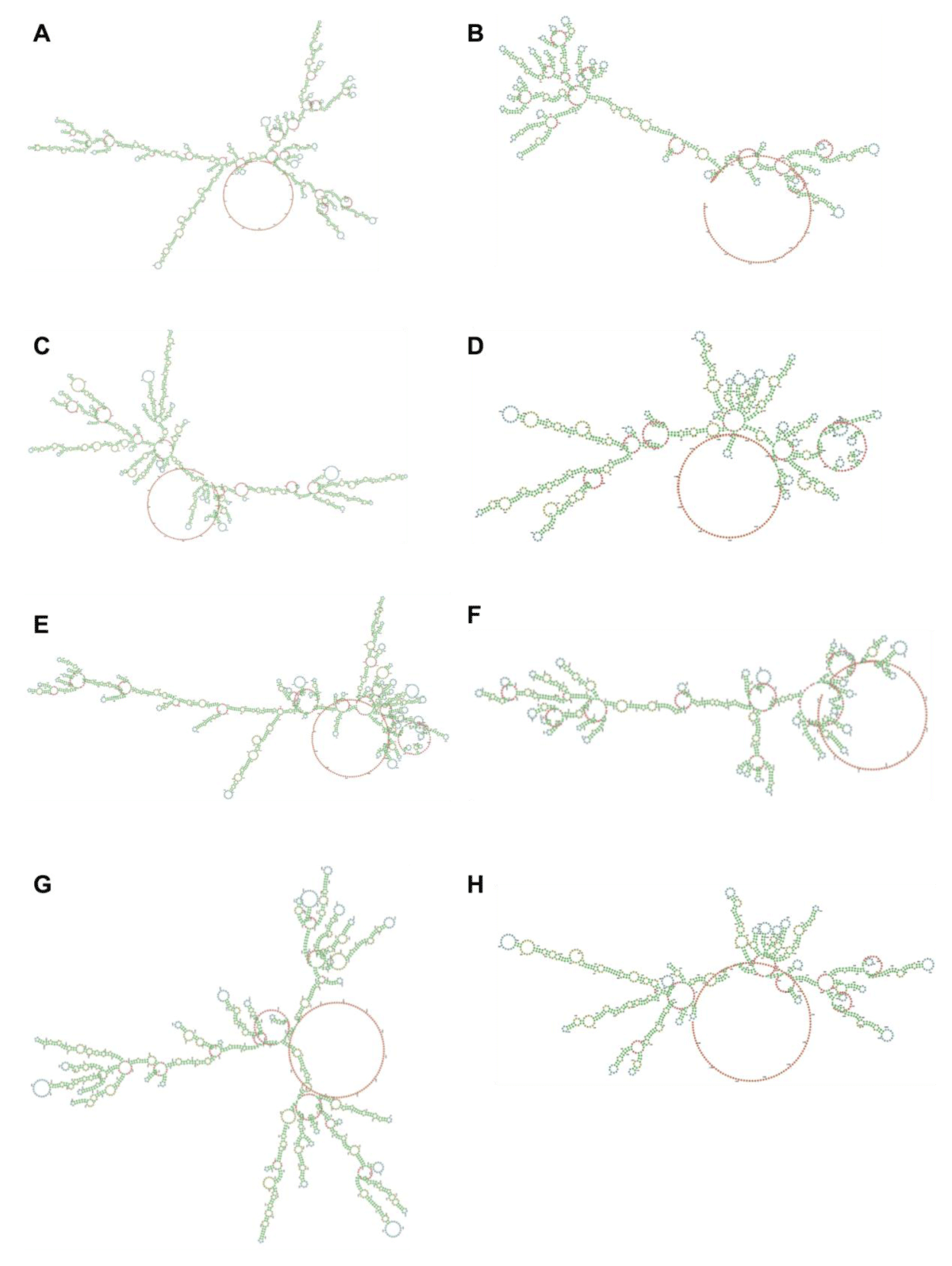
Figure S1: Predicted secondary structures of designed mRNAs. The outcome was made by LinearDesign Web Server (beta) (http://rna.baidu.com/). Nucleotides are assigned a color according to their position in the molecule. Lower-numbered nucleotides are closer to the 5′ end and are colored green. Nucleotides in the middle are colored yellow, whereas nucleotides near the 3′ end are colored red. Nucleotides are colored according to the type of structure that they are in: green, stems (canonical helices); red, multiloops (junctions); yellow, interior loops; blue, hairpin loops; orange, 5′, and 3′ unpaired region. (A) The heavy chain of mRNA-B (MFE = -629.2 kcal/mol); (B) the light chain of mRNA-B (MFE = -399.6 kcal/mol); (C) the heavy chain of mRNA-BC (MFE = -597.1 kcal/mol); (D) the light chain of mRNA-BC (MFE = -347.8 kcal/mol); (E) the heavy chain of mRNA-BU (MFE = -623.0 kcal/mol); (F) the light chain of mRNA-BU (MFE = -378.1 kcal/mol); (G) the heavy chain of mRNA-BCU (MFE = -582.7 kcal/mol); and (H) the light chain of mRNA-BCU (MFE = -346.1 kcal/mol).
MFE is the minimum free energy; the smaller the MFE, the more stable the mRNA. GC% is the ratio of guanine to cytosine; the higher the GC content, the more stable the mRNA. CAI, codon adaptation index; the CAI values range from 0 to 1, and the greater the value, the stronger the fitness.
We prepared the optimized mRNAs (Figure 3B) and investigated their in vitro expression (Figure 3C). The expression efficiency of mRNA-BC and mRNA-BU was significantly higher than that of mRNA-B, whereas the optimization of mRNA-BCU did not have a significant effect. Notably, UTR substitution or codon optimization alone significantly improved the expression levels of mRNA-B but not simultaneous optimization, which may have resulted from the synergism of the mRNA structural elements.
Furthermore, we injected 150 µg of LNP-encapsulated mRNA into mice via the caudal vein to investigate the in vivo expression efficiency and pharmacokinetic characteristics, and mice challenged with an equivalent dose of the original AET2010 were used as a control. The peak concentrations of AET2010 expressed by LNP-encapsulated mRNAs in mice were significantly higher than that obtained with the original AET2010, and the high concentrations were maintained for a longer duration (Figure 5). Among the mRNAs, mRNA-BU displayed the highest expression efficacy of AET2010 and reached a peak concentration of 581.308 µg/mL at 24 h.
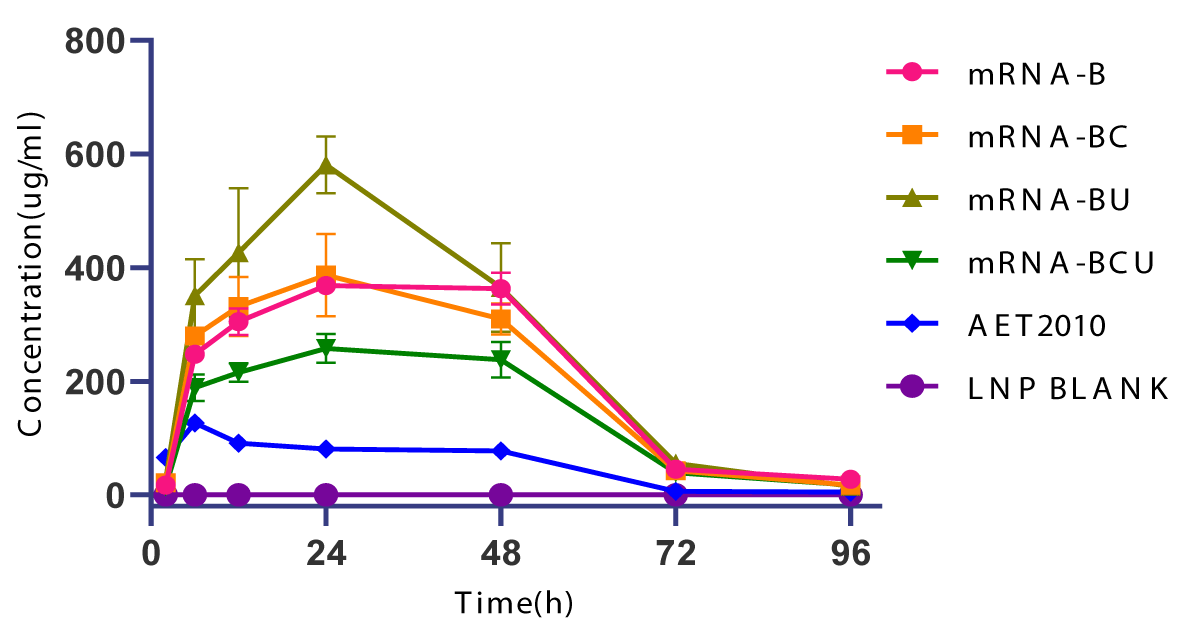
Figure 5: In vivo expression of AET2010 by lipid nanoparticle (LNP)-encapsulated mRNAs in mice. The mRNAs were packaged into LNPs using the INano™ L nanomedicine preparation system. Mice were intravenously injected with 150 µg of the original AET2010 or the mRNA-LNPs. Serum samples were collected at 2, 6, 12, 24, 48, 72, and 96 h post-administration, and the serum concentration of AET2010 was determined using enzyme-linked immunosorbent assay with recombinant TIGIT.
TIGIT is a potential target for tumor immunotherapy, and the anti-tumor efficacy of TIGIT blockade may result from the killing effect of NK and T cells on tumor cells. We obtained a candidate anti-TIGIT antibody AET2010 in a previous study and demonstrated its anti-tumor efficacy in a xenograft mouse model with an adoptive transfer of NK-92MI cells. However, the limitations of that model made it necessary to further confirm the efficacy of AET2010. As an important assessment tool for tumor immunotherapy, target-humanized mice can fully simulate target-related effector cells and be used to efficiently predict the pharmacodynamics of candidate drugs. In the present study, we used TIGIT-humanized BABL/c mice, constructed a xenograft tumor model with subcutaneous inoculation of CT26 cells, and ultimately verified the significant anti-tumor efficacy of AET2010 with a TGI rate of 50.19% (Figure 1D). This result underpinned the potential of AET2010 as a therapeutic candidate.
Theoretically, mRNAs synthesized in vitro can utilize human cells as bioreactors to produce any protein drug in vivo, which also provides a novel avenue to antibody therapy [1,28]. In cancer immunotherapy, although there are few reports about antibody-encoding mRNAs, it is still worth expecting their long-term persistence and the subsequent durable anti-tumor immune response with a low frequency and dose of administration of antibody-encoding mRNAs. In the present study, we further explored the design and optimization of mRNAs encoding AET2010. First, we designed two types of mRNAs expressing AET2010, that is the sequences encoding the light and heavy chains of the antibody were situated in one or two ORFs (mRNA-S or mRNA-B) and confirmed the high expression efficiency of mRNA-B (Figure 3C). The poor expression efficiency of mRNA-S is most likely explained by its ultralong sequence. Simultaneously, we attempted to synthesize circular mRNAs encoding the heavy chain and light chain of AET2010 separately, but the circularization efficiency was too low to achieve success (data not shown). Furthermore, based on mRNA-B, we revealed that the optimization by UTR substitution (mRNA-BU) significantly improved the in vitro and in vivo expression efficiency of AET2010 (Figure 3C and Figure 5). However, the codon usage bias optimization alone (mRNA-BC) or its combination with UTR substitution (mRNA-BCU) did not lead to significant improvement in the in vitro or in vivo expression, which may have resulted from the structures of mRNAs and their LNP encapsulation. Notably, durable expression of active AET2010 in high concentrations was observed following in vivo administration of mRNAs, which signifies that AET2010-encoding mRNAs have a potential application value. In addition, further studies need to investigate whether circular mRNAs or self-replicating mRNAs encoding AET2010 have higher expression efficiency. Based on the present findings, we plan to further investigate the strong pharmacodynamic effects of AET2010-encoding mRNAs with reduced administration frequency and dose, especially mRNA-BU.
In summary, we confirmed the anti-tumor efficacy of the candidate anti-TIGIT antibody AET2010 and obtained a candidate mRNA that efficiently encodes active AET2010 in vivo (mRNA-BU), which provides a basis for the development of novel drugs targeting TIGIT.
Author contributions
Conceptualization, P.D. and J.C.; Methodology, J.C., G.Q., and X.W.; Software, J.C.; Formal analysis, J.C., P.D., and G.Q.; Investigation, J.C. and G.Q.; Resources, J.C. and G.Q.; Data curation, J.C.; Writing—original draft preparation, J.C., G.Q., and P.D.; Writing—review and editing, Y.C., Z.Y., J.C., F.L., H.Q., and P.D.; Supervision, P.D., Y.C., Z.Y., and F.L.; Project administration, Y.C., Z.Y., P.D., and J.C. All authors have read and agreed to the submitted version of the manuscript.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
We thank Dr. Xiaofan Zhao (Beijing Institute of Biotechnology) for her assistance with the LNP-encapsulation technique. We thank Prof. Yan Zhang (Beijing Institute of Biotechnology) for the predictions of the secondary structure of mRNAs. We also thank Dr. Xiangjun Zhou (Shenzhen Yuanxing Gene-tech Co., Ltd., Shenzhen, China) for his technical support.
- Schlake T, Thran M, Fiedler K, Heidenreich R, Petsch B, Fotin-Mleczek M. mRNA: A Novel Avenue to Antibody Therapy? Mol Ther. 2019 Apr 10;27(4):773-784. doi: 10.1016/j.ymthe.2019.03.002. Epub 2019 Mar 6. PMID: 30885573; PMCID: PMC6453519.
- Trepotec Z, Geiger J, Plank C, Aneja MK, Rudolph C. Segmented poly(A) tails significantly reduce recombination of plasmid DNA without affecting mRNA translation efficiency or half-life. RNA. 2019 Apr;25(4):507-518. doi: 10.1261/rna.069286.118. Epub 2019 Jan 15. PMID: 30647100; PMCID: PMC6426288.
- Ferizi M, Aneja MK, Balmayor ER, Badieyan ZS, Mykhaylyk O, Rudolph C, Plank C. Human cellular CYBA UTR sequences increase mRNA translation without affecting the half-life of recombinant RNA transcripts. Sci Rep. 2016 Dec 15;6:39149. doi: 10.1038/srep39149. PMID: 27974853; PMCID: PMC5156912.
- Orlandini von Niessen AG, Poleganov MA, Rechner C, Plaschke A, Kranz LM, Fesser S, Diken M, Löwer M, Vallazza B, Beissert T, Bukur V, Kuhn AN, Türeci Ö, Sahin U. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3' UTRs Identified by Cellular Library Screening. Mol Ther. 2019 Apr 10;27(4):824-836. doi: 10.1016/j.ymthe.2018.12.011. Epub 2018 Dec 18. PMID: 30638957; PMCID: PMC6453560.
- Bae H, Coller J. Codon optimality-mediated mRNA degradation: Linking translational elongation to mRNA stability. Mol Cell. 2022 Apr 21;82(8):1467-1476. doi: 10.1016/j.molcel.2022.03.032. PMID: 35452615.
- Hanson G, Coller J. Codon optimality, bias and usage in translation and mRNA decay. Nat Rev Mol Cell Biol. 2018 Jan;19(1):20-30. doi: 10.1038/nrm.2017.91. Epub 2017 Oct 11. PMID: 29018283; PMCID: PMC6594389.
- Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, Coller J. Codon optimality is a major determinant of mRNA stability. Cell. 2015 Mar 12;160(6):1111-24. doi: 10.1016/j.cell.2015.02.029. PMID: 25768907; PMCID: PMC4359748.
- Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, Felgner PL. Direct gene transfer into mouse muscle in vivo. Science. 1990 Mar 23;247(4949 Pt 1):1465-8. doi: 10.1126/science.1690918. PMID: 1690918.
- Jirikowski GF, Sanna PP, Maciejewski-Lenoir D, Bloom FE. Reversal of diabetes insipidus in Brattleboro rats: intrahypothalamic injection of vasopressin mRNA. Science. 1992 Feb 21;255(5047):996-8. doi: 10.1126/science.1546298. PMID: 1546298.
- Beck JD, Reidenbach D, Salomon N, Sahin U, Türeci Ö, Vormehr M, Kranz LM. mRNA therapeutics in cancer immunotherapy. Mol Cancer. 2021 Apr 15;20(1):69. doi: 10.1186/s12943-021-01348-0. PMID: 33858437; PMCID: PMC8047518.
- Pardi N, Secreto AJ, Shan X, Debonera F, Glover J, Yi Y, Muramatsu H, Ni H, Mui BL, Tam YK, Shaheen F, Collman RG, Karikó K, Danet-Desnoyers GA, Madden TD, Hope MJ, Weissman D. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat Commun. 2017 Mar 2;8:14630. doi: 10.1038/ncomms14630. PMID: 28251988; PMCID: PMC5337964.
- Alfagih IM, Aldosari B, AlQuadeib B, Almurshedi A, Alfagih MM. Nanoparticles as Adjuvants and Nanodelivery Systems for mRNA-Based Vaccines. Pharmaceutics. 2020 Dec 30;13(1):45. doi: 10.3390/pharmaceutics13010045. PMID: 33396817; PMCID: PMC7823281.
- Paunovska K, Loughrey D, Dahlman JE. Drug delivery systems for RNA therapeutics. Nat Rev Genet. 2022 May;23(5):265-280. doi: 10.1038/s41576-021-00439-4. Epub 2022 Jan 4. PMID: 34983972; PMCID: PMC8724758.
- Kiaie SH, Majidi Zolbanin N, Ahmadi A, Bagherifar R, Valizadeh H, Kashanchi F, Jafari R. Recent advances in mRNA-LNP therapeutics: immunological and pharmacological aspects. J Nanobiotechnology. 2022 Jun 14;20(1):276. doi: 10.1186/s12951-022-01478-7. PMID: 35701851; PMCID: PMC9194786.
- Meo SA, Bukhari IA, Akram J, Meo AS, Klonoff DC. COVID-19 vaccines: comparison of biological, pharmacological characteristics and adverse effects of Pfizer/BioNTech and Moderna Vaccines. Eur Rev Med Pharmacol Sci. 2021 Feb;25(3):1663-1669. doi: 10.26355/eurrev_202102_24877. PMID: 33629336.
- To KKW, Cho WCS. An overview of rational design of mRNA-based therapeutics and vaccines. Expert Opin Drug Discov. 2021 Nov;16(11):1307-1317. doi: 10.1080/17460441.2021.1935859. Epub 2021 Jul 19. PMID: 34058918.
- August A, Attarwala HZ, Himansu S, Kalidindi S, Lu S, Pajon R, Han S, Lecerf JM, Tomassini JE, Hard M, Ptaszek LM, Crowe JE, Zaks T. A phase 1 trial of lipid-encapsulated mRNA encoding a monoclonal antibody with neutralizing activity against Chikungunya virus. Nat Med. 2021 Dec;27(12):2224-2233. doi: 10.1038/s41591-021-01573-6. Epub 2021 Dec 9. Erratum in: Nat Med. 2022 May;28(5):1095-1096. PMID: 34887572; PMCID: PMC8674127.
- Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, Levine Z, Beiman M, Dassa L, Achdout H, Stern-Ginossar N, Tsukerman P, Jonjic S, Mandelboim O. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A. 2009 Oct 20;106(42):17858-63. doi: 10.1073/pnas.0903474106. Epub 2009 Oct 7. PMID: 19815499; PMCID: PMC2764881.
- Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. 2020 Sep;8(2):e000957. doi: 10.1136/jitc-2020-000957. PMID: 32900861; PMCID: PMC7477968.
- Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, Eaton DL, Grogan JL. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014 Dec 8;26(6):923-937. doi: 10.1016/j.ccell.2014.10.018. Epub 2014 Nov 26. PMID: 25465800.
- Han D, Xu Y, Zhao X, Mao Y, Kang Q, Wen W, Yu X, Xu L, Liu F, Zhang M, Cui J, Wang Z, Yang Z, Du P, Qin W. A novel human anti-TIGIT monoclonal antibody with excellent function in eliciting NK cell-mediated antitumor immunity. Biochem Biophys Res Commun. 2021 Jan 1;534:134-140. doi: 10.1016/j.bbrc.2020.12.013. Epub 2020 Dec 16. PMID: 33341068.
- Clarke TF 4th, Clark PL. Rare codons cluster. PLoS One. 2008;3(10):e3412. doi: 10.1371/journal.pone.0003412. Epub 2008 Oct 15. PMID: 18923675; PMCID: PMC2565806.
- Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010 Mar 15;11:129. doi: 10.1186/1471-2105-11-129. PMID: 20230624; PMCID: PMC2984261.
- Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004 Jul;22(7):346-53. doi: 10.1016/j.tibtech.2004.04.006. PMID: 15245907.
- Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, McCullough MP, Chappell JD, Denison MR, Stevens LJ, Pruijssers AJ, McDermott A, Flach B, Doria-Rose NA, Corbett KS, Morabito KM, O'Dell S, Schmidt SD, Swanson PA 2nd, Padilla M, Mascola JR, Neuzil KM, Bennett H, Sun W, Peters E, Makowski M, Albert J, Cross K, Buchanan W, Pikaart-Tautges R, Ledgerwood JE, Graham BS, Beigel JH; mRNA-1273 Study Group. An mRNA Vaccine against SARS-CoV-2 - Preliminary Report. N Engl J Med. 2020 Nov 12;383(20):1920-1931. doi: 10.1056/NEJMoa2022483. Epub 2020 Jul 14. PMID: 32663912; PMCID: PMC7377258.
- Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen IJ, Morris DR, Seelig G. Human 5' UTR design and variant effect prediction from a massively parallel translation assay. Nat Biotechnol. 2019 Jul;37(7):803-809. doi: 10.1038/s41587-019-0164-5. Epub 2019 Jul 1. PMID: 31267113; PMCID: PMC7100133.
- Linares-Fernández S, Moreno J, Lambert E, Mercier-Gouy P, Vachez L, Verrier B, Exposito JY. Combining an optimized mRNA template with a double purification process allows strong expression of in vitro transcribed mRNA. Mol Ther Nucleic Acids. 2021 Dec 3;26:945-956. doi: 10.1016/j.omtn.2021.10.007. Epub 2021 Oct 19. PMID: 34692232; PMCID: PMC8523304.
- Van Hoecke L, Roose K. How mRNA therapeutics are entering the monoclonal antibody field. J Transl Med. 2019 Feb 22;17(1):54. doi: 10.1186/s12967-019-1804-8. PMID: 30795778; PMCID: PMC6387507.